GC-MS/MS法测定不同级别乳酸中残留溶剂的含量
2022-09-13张悦赵勇郑金凤刘雁鸣谢莹莹湖南省药品检验检测研究院长沙410001国家药品监督管理局药用辅料工程技术研究重点实验室长沙410001
张悦,赵勇,郑金凤,刘雁鸣,谢莹莹*(1.湖南省药品检验检测研究院,长沙 410001;2.国家药品监督管理局药用辅料工程技术研究重点实验室,长沙 410001)
乳酸(lactic acid)又名2-羟基丙酸,是一种天然存在的重要有机酸,作为消毒防腐剂、pH调节剂、助溶剂等,可用于肌内注射、静脉注射、皮下注射、口服糖浆、片剂、外用和阴道用制剂[1-2]。
乳酸的微生物发酵生产过程中可能产生甲醇、乙醇等有机副产物,采用酯化水解法也可能使用甲醇和乙醇与乳酸成酯[3-4]。这些溶剂的残留会对人体健康造成危害,控制其残留量对保证药品质量和用药安全有重要意义。目前欧洲药典(EP10.0)[5]规定了注射用乳酸中甲醇的限度(不得过50 μg·g-1),而我国尚未见乳酸中残留溶剂检测方面的相关报道。为考察国内乳酸中甲醇、乙醇残留的整体情况,本研究根据《人用药品注册技术要求国际协调会质量部分》(ICH)[6]的指导原则(甲醇每日允许暴露量30.0 μg·g-1,乙醇每日允许暴露量50.0 μg·g-1)和《中国药典》2020年版四部[7]中的相关规定,并参考相关文献报道[8-10],拟建立GC-MS/MS法测定乳酸中甲醇、乙醇的含量,并评价国内市场收集的33批乳酸的总体质量。
1 仪器与试药
1.1 仪器
8890-7010B GC-MS/MS色谱仪(美国安捷伦公司);MS105DU 电子天平(瑞士Mettler-Toledo公司,精度为十万分之一);VF-WAX ms毛细管柱(30 m×0.25 mm,0.25 μm)(美国安捷伦公司);固定液为聚乙二醇(PEG-20 M)。
1.2 试药
甲醇(纯度≥99.99%,美国霍尼韦尔公司,批号:12G1H);乙醇(纯度≥99.9%,德国CNW Technologies GmbH公司,批号:E5450210)。33批乳酸样品包括12批原料药级、13批药用辅料、5批食品级、2批工业级及1批进口高纯级,详细信息见表1。
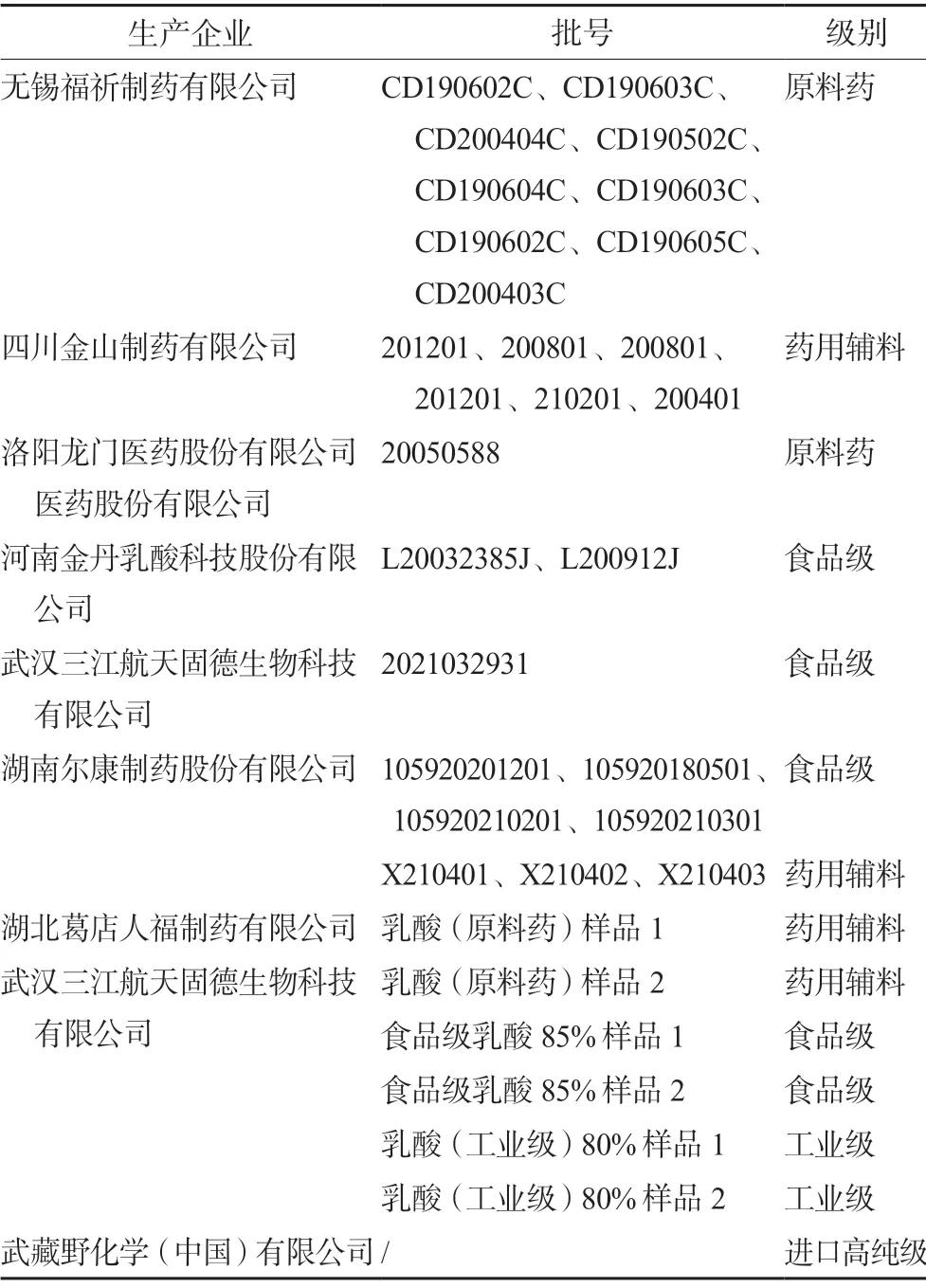
表1 乳酸样品信息Tab 1 Lactic acid sample information
2 方法与结果
2.1 溶液的配制
2.1.1 对照品溶液的制备 分别取甲醇、乙醇适量,精密称定,用水溶解并稀释成10 mg·mL-1的溶液,作为标准储备液。再分别量取适量标准储备液,用水配制成质量浓度为0、5、10、50、100、150 μg·mL-1的系列对照品溶液。
2.1.2 供试品溶液的制备 取乳酸0.5 g,精密称定,置20 mL 顶空瓶中,精密加入5 mL 水溶解,即得。
2.2 色谱条件
程序升温:初温50℃,保持4 min,以30℃·min-1的升温速率升至200℃,保持2 min;进样口温度220℃;载气为高纯He(纯度>99.999%);流速2.0 mL·min-1,分流进样,分流比5∶1;进样量1.0 mL;顶空瓶平衡温度80℃,平衡时间30 min,定量环温度90℃,传输线温度100℃。
2.3 质谱条件
采用电子轰击离子源(EI),电子能量70 eV,离子源温度230℃,溶剂延迟0.5 min,采用多重反应监测模式(MRM)扫描。甲醇:定量离子对为m/z32.1>30.1,碰撞能量(CE)5 eV;定性离子对为m/z32.1>28.1,碰撞能量5 eV。乙醇:定量离子对为m/z46.1>45.1,碰撞能量5 eV;定性离子对为m/z46.1>31.1,碰撞能量5 eV。
2.4 系统适用性试验
精密量取“2.1”项下空白溶液(水)、对照品溶液和供试品溶液各5 mL,注入GC-MS/MS仪,记录色谱图,结果见图1,空白溶液无干扰,各杂质能有效分离。
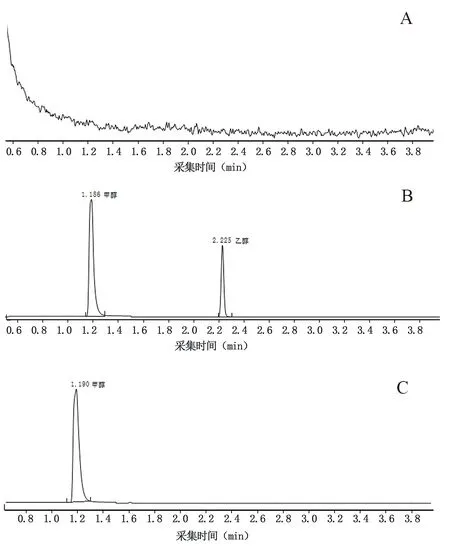
图1 系统适用性色谱图Fig 1 Chromatogram of specificity test
2.5 线性关系、检出限与定量限
精密量取甲醇、乙醇标准储备液,用水定量稀释制成0、5、10、50、100、150 μg·mL-1系列对照品溶液,分别进样,记录峰面积。以质量浓度(x,μg·mL-1)为横坐标,以峰面积(y)为纵坐标,进行线性回归,得回归方程:y甲醇=1.82x+0.65,r=0.9999;y乙醇=3.99x-0.27,r=0.9997。甲醇、乙醇在0~150 μg·mL-1内与峰面积呈良好的线性关系。采用信噪比S/N≈3计算检出限,得甲醇、乙醇的检出限分别为9.85、11.60 ng·mL-1;S/N≈10计算定量限,得甲醇、乙醇的定量限分别为32.83、38.67 ng·mL-1。
2.6 重复性试验
取样品(批号:CD190602C)0.5 g,置顶空瓶中,加入“2.1.1”项下对照品溶液(甲醇49.2 μg·mL-1,乙醇50.1 μg·mL-1)5 mL,平行制备6份,进样测定,记录色谱峰面积,计算甲醇、乙醇含量。结果甲醇平均含量为45.7 μg·mL-1,RSD为4.6%;乙醇平均含量为45.2 μg·mL-1,RSD为5.7%。说明本方法重复性好。
2.7 稳定性试验
取 样 品(批 号:CD190602C)5 g,置50 mL量瓶中,加对照品溶液(甲醇、乙醇:50 μg·mL-1)50 mL稀释并定容至刻度,再精密量取5 mL,置顶空瓶中,于0、2、4、8、12、20 h进样,记录峰面积。结果供试品溶液在20 h内甲醇、乙醇峰面积的RSD分别为4.5%、5.3%(n=6),表明方法稳定性良好。
2.8 回收试验
取样品(批号:CD190602C)约0.5 g,共6份,置顶空瓶中,精密加入对照品溶液(甲醇4.987 μg·mL-1,乙 醇5.132 μg·mL-1)5 mL,进样测定。结果甲醇、乙醇平均回收率分别为93.5%(RSD=4.0%)、89.8%(RSD=4.3%),说明本法准确度好。
2.9 样品测定
取33批乳酸样品,按“2.1”项下方法制备,平行制备两份,进样测定,计算样品中甲醇、乙醇含量,结果有2批次检测出残留溶剂,四川金川制药有限公司200801批次的样品检出乙醇9.4 μg·mL-1,武藏野化学(中国)有限公司的样品检出甲醇8.7 μg·mL-1。
3 讨论
3.1 色谱柱的选择
选择质谱专用毛细管色谱柱DB-1 ms、HP-5 ms、DB-624 ms和VF-WAX ms进行试验。结果显示VF-WAX ms毛细管柱进行分离时,分离效果最好,且灵敏度较高,峰形最尖锐、对称,故选用VF-WAX ms毛细管柱为色谱柱。
3.2 质谱条件的选择
取10 μg·mL-1的甲醇、乙醇溶液进行MS1 Scan 模式全扫描,扫描范围10~200 amu,通过谱库检索确定各成分,并选择丰度最高的离子作为母离子(m/z32.1作为甲醇母离子,m/z46.1作为乙醇母离子)。再对母离子进行Product ion扫描,确定各产物离子的时间片断和驻留时间,选择丰度最高的特征碎片离子作为子离子。设定不同碰撞池电压的运行序列(CE:3、5、8、10 V),优化子离子的最佳碰撞能量,建立MRM方法。
3.3 平衡温度和平衡时间的考察
顶空气体的浓度随着平衡温度的升高而升高,当瓶内处于气液平衡后,各组分响应值趋于稳定[11-13]。本研究考察了不同温度80、90、100、110℃下各组分的响应情况。发现甲醇、乙醇响应值随平衡温度升高而增大,但温度升至90℃以后,响应值变化不明显。考虑到平衡温度太高,顶空瓶气密性变差,会影响测定结果的稳定性,故选择90℃作为最佳的平衡温度。在平衡温度90℃条件下,对不同平衡时间20、30、40、50 min的平衡效果进行考察,发现甲醇、乙醇响应值随着时间的增加而增加,当时间超过30 min后,响应值变化较小,故最终选择30 min作为最佳的平衡时间。
3.4 国内市场中不同级别乳酸质量情况
本研究结果显示,33批乳酸样品中,仅1批样品检出乙醇(为药用辅料级),1批样品检出甲醇(为进口高纯级),其他样品均未检出残留溶剂。《中国药典》2020年版四部通则0861残留溶剂测定法规定乙醇限度为0.5%,甲醇限度为0.3%;EP10.0规定注射用乳酸控制甲醇不得过50 μg·g-1。本研究33批次乳酸中的乙醇和甲醇残留量均远低于上述限度。说明国内市场中原料药、药用辅料、食品级、工业级、进口高纯级等不同级别乳酸整体工艺控制良好,甲醇、乙醇残留量较低。
甲醇是ICH中第二类溶剂,是非遗传毒性动物致癌或可能导致其他不可逆毒性神经毒性或致畸形的试剂,为应限制的溶剂。本次研究收集到1批进口高纯级样品,且在这批样品中检出少量甲醇,故应多关注高纯级样品的溶剂残留情况。另外研究中还发现,有药品生产企业混用不同级别乳酸的情况,如将食品级(2021032931)乳酸当作药用辅料级用于药品生产中。故监管部门应多关注乳酸的混用情况,特别注意高纯级乳酸用于药品或食品市场中的溶剂残留情况。
