阿卡波糖口服固体制剂含量测定方法的优化
2022-09-13石笑弋易必新李昌亮李帅刘雁鸣兰文李健和湖南省药品检验检测研究院长沙4000国家药品监督管理局药用辅料工程技术研究重点实验室长沙4000中南大学湘雅二医院药学部长沙400
石笑弋,易必新*,李昌亮,2,李帅,2,刘雁鸣,2,兰文,2,李健和(.湖南省药品检验检测研究院,长沙 4000;2.国家药品监督管理局药用辅料工程技术研究重点实验室,长沙 4000;.中南大学湘雅二医院药学部,长沙 400)
阿卡波糖是德国拜耳公司于20 世纪80年代从一种游离放线菌发酵分离得到的产物,由4 个单糖构成的寡糖,是世界上第一个α
-葡萄糖糖苷酶抑制剂类的口服降糖药,主要用于2 型糖尿病的治疗,可通过抑制淀粉酶来减少葡萄糖的吸收,更适合于降低以碳水化合物为主食的亚洲糖尿病患者的血糖。阿卡波糖口服固体制剂自1994年在中国批准上市以来,不良反应相对较小,市场占有率较高,应用前景较广。目前,国内现有阿卡波糖及其制剂质量标准收载于《中国药典》2020年版二部,其他相关质量标准及文献中阿卡波糖含量测定的分析方法多采用HPLC 法,检测器普遍应用紫外或示差折光检测器,亦有利用电喷雾检测器等新型检测器检测,色谱柱大多使用氨基柱,流动相均采用大量有机试剂。现有方法大多存在受环境影响基线波动较大、灵敏度降低、柱效下降快或普及率不高等问题。因此,对阿卡波糖片及胶囊的含量测定方法进行改进十分必要。本文采用SCX 色谱柱,优化了阿卡波糖口服固体制剂中阿卡波糖含量测定的方法,现报道如下。
1 仪器与试药
赛默飞Dionex UltiMate 3000 高效液相色谱仪、PDA 紫外检测器;十万分之一XP205 电子分析天平(Mettler Toledo)。阿卡波糖对照品(批号:100808-201905,含量:99.4%,中国食品药品检定研究院,水分:3.45%)。阿卡波糖原料药(杭州中美华东制药有限公司,批号:1909156AK)。1个厂家的阿卡波糖胶囊及6 个厂家的阿卡波糖片共165 批次样品。磷酸二氢钾、氢氧化钠、盐酸等试剂为分析纯(国药集团化学试剂有限公司);纯水(Thermo Scientific GenPure 超纯水机制备)。
2 方法与结果
2.1 色谱条件
色谱柱Welch Ultimate XB-SCX 柱(4.6 mm×250 mm,3 μm),流动相0.02 mol·L磷酸二氢钾溶液,流速1.0 mL·min,柱温40℃,检测波长210 nm,进样量10 μL。
2.2 溶液的配制
2.2.1 供试品溶液的配制 取本品20 片(或20粒内容物),精密称定,研细,精密称取适量(约相当于阿卡波糖50 mg),置50 mL 量瓶中,加水适量,超声使溶解并稀释至刻度,摇匀,滤过,取续滤液作为供试品溶液。
2.2.2 对照品溶液的配制 取阿卡波糖对照品适量,加水制成每1 mL 中约含1 mg 的溶液作为对照品溶液。
2.2.3 阴性样品溶液的配制 按处方比例及制备工艺,制备缺阿卡波糖的样品,按“2.2.1”项下方法制备阴性样品溶液。
2.2.4 系统适用性溶液的配制 取阿卡波糖原料药约200 mg,置10 mL 量瓶中,加少量水使溶解,加0.1 mol·L氢氧化钠溶液1 mL,混匀,室温放置1 h,加0.1 mol·L盐酸溶液1 mL,加水稀释至刻度,摇匀,作为系统适用性溶液。
2.3 系统适用性试验
精密量取“2.2”项下阴性样品溶液、系统适用性溶液及供试品溶液各10 μL,进样测定,结果主峰与相邻杂质峰均有效分离,以阿卡波糖色谱峰计,拖尾因子小于1.2,理论板数大于5000。溶剂、辅料对测定均无干扰,见图1。
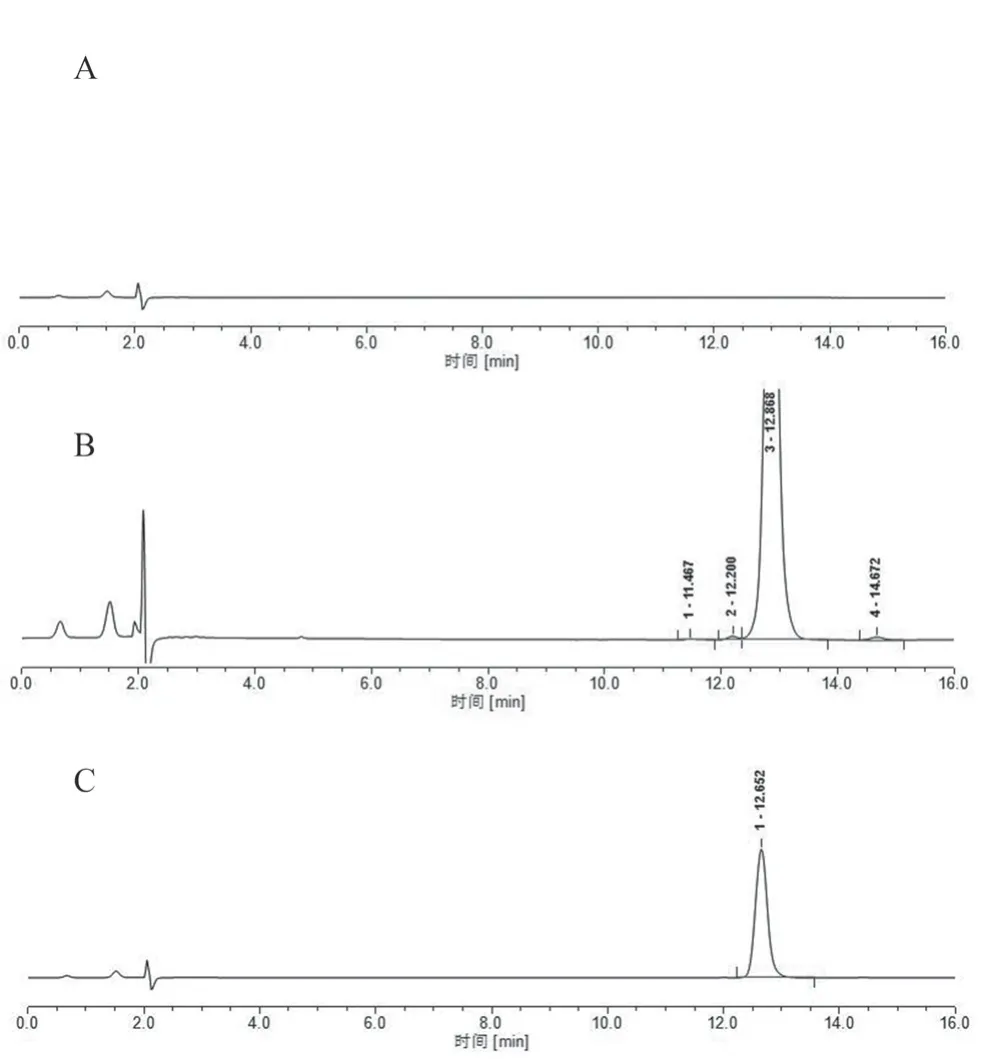
图1 系统适用性试验HPLC 图Fig 1 HPLC chromatogram of system suitability test
2.4 线性范围、检测限与定量限
精密称取阿卡波糖对照品215.19 mg,置50 mL 量瓶中,加水适量,超声使溶解并稀释至刻度,摇匀,作为线性溶液①,取该线性溶液6、3、2.5、2、0.5、0.2 mL,分别置10 mL 量瓶中,加水稀释至刻度,摇匀,得到质量浓度为2.478、1.239、1.032、0.8260、0.2065、0.082 60 mg·mL的线性溶液②~⑦。进样测定,记录色谱图。以质量浓度(X
)为横坐标和峰面积(Y
)为纵坐标进行线性回归,得阿卡波糖回归方程为Y
=2.529×10X
-244.2,相关系数为1.000,线性范围为0.082 60 ~4.130 mg·mL;并根据信噪比S/N
≈ 3 确定检测限(LOD),S/N
≈ 10 确定定量限(LOQ),结果检测限为6.8 μg·mL,定量限为22 μg·mL。2.5 精密度试验
取“2.4”项下质量浓度约为1.0 mg·mL的对照品溶液,连续进样6 次,记录保留时间与峰面积。结果阿卡波糖峰面积的RSD
<1.5%,保留时间的RSD
均<1.0%,表明仪器精密度良好。2.6 重复性试验
取同一批样品(阿卡波糖片,批号:210301,规格:50 mg)6 份,按“2.2.1”项下方法制备并测定,结果含量平均值为100.11%,RSD
为0.70%,表明方法重复性良好。2.7 溶液稳定性试验
取“2.5”项下溶液室温避光放置,分别于0、2、4、8、12、16、20 及24 h 进样考察,阿卡波糖的峰面积RSD
均<2.0%,表明溶液在24 h 内稳定性良好。2.8 回收试验
取阴性样品,精密加入阿卡波糖对照品,制成浓度相当于阿卡波糖含量100%水平的供试溶液共6 份,进样分析,记录色谱图,按外标法以峰面积计算阿卡波糖的含量,结果平均回收率为99.41%,RSD
为0.32%(n
=6)。2.9 耐用性试验
通过调节流速及柱温,考察阿卡波糖峰的变化情况。结果阿卡波糖峰的理论板数均大于5000,拖尾因子均小于1.2,与相邻杂质峰均能有效分离。
2.10 样品测定
取165 批次样品,按“2.2.1”项下方法制备供试品溶液,按“2.1”项下色谱条件进行测定,按外标法以峰面积计算,并将含量测定结果与《中国药典》2020年版二部收载的采用氨基柱测定的结果进行比较,采用SPSS 统计软件对两种方法含量测定测结果进行配对样品t
检验。结果相关系数r
=0.542,P
<0.001,认为两配对变量有相关关系;t
=-3.263,d
=164,双尾检验概率P
=0.001,表明两种检验方法所测结果具有显著性差异,结果见表1。表1 样品中阿卡波糖的含量结果( =165)
Tab 1 Content of acarbose in the sample ( =165)
含量均值的标准差/%药典(采用NH2 色谱柱)98.811.12640.0877本文(采用SCX 色谱柱)99.091.21600.0946测定方法含量均值/%标准差/%
3 讨论
《中国药典》2020年版二部是采用HPLC 法测定阿卡波糖的含量。采用氨基色谱柱,以乙腈-磷酸盐缓冲液(75∶25)为流动相,流速2 mL·min,柱温35℃,检测波长 210 nm。该方法存在很多缺点,严重影响了方法的重现,并造成应用困难:① 相邻降解杂质Ⅰ峰与主峰的分离效果不佳,只有系统适用性中规定杂质Ⅰ的峰高(Hp)与杂质Ⅰ和阿卡波糖两峰之间的峰谷(Hv)之比Hp/Hv不得低于2.0 的要求;② 氨基柱耐用性较差,键合相容易流失,最多使用150 次左右,柱效严重降低,色谱峰峰形变差,影响准确定量;③ 流动相中有机试剂毒性大,流速大,检测成本高。因此,有必要优化阿卡波糖含量测定的分析方法,改善方法的重现性和可应用性,提高药品质量控制水平。目前尚未见采用SCX 色谱柱HPLC 法测定阿卡波糖含量的报道。
3.1 流动相的选择
阿卡波糖在水中极易溶解,在乙腈中不溶。本文采用水为溶剂,0.02 mol·L磷酸二氢钾溶液为流动相,能解决《中国药典》2020年版二部方法中因溶剂与流动相中有机相比例差异对灵敏度、出峰时间及峰形等的影响。
3.2 色谱条件的优化
本文以0.02 mol·L磷酸二氢钾溶液为流动相,流速1.0 mL·min,柱温40℃的色谱条件,采用SCX 色谱柱(4.6 mm×250 mm,3 μm),系统适用性要求均能达到。SCX 色谱柱是一种典型的阳离子交换柱,以磺酸基强阳离子硅胶为键合相,用于碱性、水溶的化合物的分离。SCX 色谱柱较氨基色谱柱的固定相稳定,不易产生柱流失,在使用约300 次后,组分仍然能保持良好的峰形,对称性好,拖尾因子小,分离度与理论板数能满足系统适用性要求。
3.3 小结
本文采用SCX 色谱柱的HPLC 法测得的阿卡波糖口服固体制剂含量结果准确、方法可靠,与《中国药典》2020年版收载的方法相比,该方法毒性小、成本低、专属性强、色谱柱耐用性好,具备一定的实用性。
