糖酵解与线粒体功能障碍的关系及其在肝脏疾病中的潜在价值
2022-09-08颜耿杰苏会吉陈含笑班少群韦艾凌毛德文龙富立
颜耿杰, 林 镛, 苏会吉, 陈含笑, 班少群, 韦艾凌,, 毛德文, 龙富立
1 广西中医药大学 研究生院, 南宁 530200; 2 广西中医药大学第一附属医院 肝病二区, 南宁 530023
肝脏是控制全身能量代谢的主要器官,包括葡萄糖、脂肪酸和氨基酸代谢,在体内营养平衡中承担核心作用。葡萄糖和脂肪酸代谢主要发生在线粒体中。线粒体在肝细胞中数量非常庞大,占细胞体积的18%~20%,且更集中于三磷酸腺苷(adenosine triphosphate,ATP)利用位点附近[1],同时在能量代谢和细胞稳态中发挥多种作用,包括细胞呼吸、氧化磷酸化、活性氧(reactive oxygen species,ROS)平衡和细胞死亡的调节等[2]。然而,在各种不利条件下,葡萄糖和脂质代谢功能受到破坏,如糖酵解、糖异生、脂肪生成和脂肪酸氧化,这些代谢过程均在肝脏中进行。其中,糖酵解对肝脏疾病发生发展的影响近年受到越来越多的研究关注。
1 糖酵解促进肝脏疾病进展
糖酵解是指葡萄糖被催化为丙酮酸,并提供2个还原型烟酰胺腺嘌呤二核苷酸和2个ATP的过程。丙酮酸可被丙酮酸脱氢酶(pyruvate dehydrogenase,PDH)氧化为乙酰辅酶A,或者通过线粒体中的丙酮酸羧化酶转化为草酰乙酸。在无氧条件下的丙酮酸通过乳酸脱氢酶(lactate dehydrogenase,LDH)还原为乳酸盐,或通过丙酮酸脱羧酶脱羧为乙醛。当肝脏处于病理状态时,能量代谢将优先从氧化磷酸化转换为糖酵解,其结果是部分丙酮酸被转换为乳酸盐[3]。研究[4]表明,糖酵解过程改变的机制可促进非酒精性脂肪性肝病(NAFLD)进展为非酒精性脂肪性肝炎(NASH),最终进展为肝硬化和肝细胞癌(HCC)的过程与氧化还原失衡和线粒体功能障碍相关。高脂饮食可抑制PDH的活性,并降低肝线粒体清除过氧化物的能力,导致氧化应激和NAFLD的进展。另有研究[5]发现,PDH激酶同工酶4(PDK4)可抑制NASH中PDH的活性,当PDK4在NASH小鼠肝脏中缺乏时,肝脂肪变性可显著改善。综上所述,PDH的抑制将破坏氧化还原平衡并促进肝脏疾病的进展,并且PDH的激活可改善氧化还原失衡和脂肪变性。此外,糖酵解活性的增强可促进线粒体ROS的产生,使对氧化还原失衡敏感的M2型丙酮酸激酶(recombinant pyruvate kinase isozymes M2,PKM2)转移至细胞核中,从而促进IL-6和IL-1β的产生,进而引发炎症[6]。反之,ROS又可抑制线粒体内膜上的呼吸链酶,使甘油醛-3-磷酸脱氢酶和膜钠通道失活,诱导肝细胞损伤,进一步加重脂质过氧化,导致细胞因子产生和脂质积累,并促进肝脏炎症和纤维化。而由异常糖酵解引起的线粒体功能障碍与ROS产生轴之间的恶性循环可促进肝细胞凋亡并向肝硬化和HCC进展[7-8]。上述发现均提示,糖酵解异常可促进肝脏疾病的发生发展。
2 肝脏疾病增强糖酵解
糖酵解对肝脏疾病进展具有促进作用,而肝脏疾病中的病理状态也影响糖酵解过程。由于代偿线粒体中的ATP产生缺陷,糖酵解将随着肝脏疾病严重程度加重而得到不同程度的增强。研究[9]发现,肝硬化患者肝脏中的糖酵解酶,例如己糖激酶(hexokinase 2,HK2)、醛缩酶A和PKM2的基因表达较正常肝脏明显上调,并且这些基因的表达与HCC的发生风险相关。Li等[10]研究发现HCC中存在有氧糖酵解,且HCC中癌细胞的有氧糖酵解程度得到增强,即使在有氧条件下也优先将葡萄糖代谢成乳酸。上述发现均与氧化磷酸化到糖酵解代谢的转变有关,氧化磷酸化作为主要的能量产生过程,当线粒体功能障碍时,为了解决氧化磷酸化受损导致的ATP生成失衡,糖酵解速率代偿性升高,从而增强葡萄糖消耗和乳酸产生。当葡萄糖水平升高时,相对较高的糖酵解速率能够产生比氧化磷酸化更多的能量[11]。在大多数正常细胞中,线粒体氧化磷酸化和糖酵解产生的ATP分别约为90%和10%,而癌细胞依靠有氧糖酵解提供高达60%的ATP消耗[12],并且癌细胞对ATP的需求越大,糖酵解产生的乳酸在细胞中积累得越多。研究[13-14]表明,癌细胞通过LDH-A增强糖酵解产生乳酸以防止其在缺氧环境中发生细胞凋亡,因此有部分学者认为,升高的乳酸有利于癌细胞逃避免疫,并为癌症转移提供酸性肿瘤微环境。总而言之,糖酵解在不同肝脏疾病中的活性增强,并与线粒体功能障碍密切相关,这有助于弥补由氧化磷酸化受损而导致的能量缺陷,因此或可通过靶向调节有氧糖酵解中的关键因素,如抑制酶HK2、 PKM2或其他调节途径,寻求肝脏疾病的潜在治疗新方法。
3 糖酵解在肝脏疾病治疗中的应用前景
3.1 NAFLD 在NAFLD中,肝细胞的许多代谢途径发生显著变化,如糖酵解增强、乳酸生成、三羧酸循环、酮体生成减少、线粒体呼吸和ATP合成减少,这些代谢途径之间存在着密切而复杂的相互作用。其中,糖酵解中的一些相关代谢途径可能成为NAFLD潜在的治疗靶点。最近的研究[15]发现,高脂饮食可以通过LKB1-AMPK途径下调香叶基香叶基二磷酸合酶(geranylgeranyl pyrophosphate synthase,GGPPS)的表达来增强糖酵解,并导致小鼠原代肝细胞分泌的炎症因子增加。此外,使用2-脱氧-D-葡萄糖抑制糖酵解可显著缓解小鼠由于GGPPS缺失而导致的肝脏炎症和纤维化。Kors等[16]研究发现,敲除小鼠肝脏富含亮氨酸重复序列的家族蛋白X1可以抑制糖酵解,并导致脂肪酸氧化增加和脂肪变性减少,或可作为NAFLD的新型治疗靶点。Shannon等研究[17]发现,肥胖小鼠中PDH复合物的活性增强伴随着PDK4活性降低和PDH磷酸酶同工酶2活性增强,而吡格列酮可使PDH复合物的活性正常化,并抑制丙酮酸进入三羧酸循环中代谢。丙酮酸盐的另一种代谢途径是乳酸的产生,在生理条件下,乳酸主要作为心脏、骨骼肌和大脑的能量来源,而肝细胞可将过量的血浆乳酸转化为葡萄糖,然后带回血浆。p300/CBP相关因子介导的 LDH-B 的高乙酰化水平可降低肝细胞代谢乳酸盐的能力,并导致乳酸盐积累,是NAFLD中乳酸盐积累的主要原因之一,增加的乳酸不仅加重肝脂肪变性,还可以通过降低组蛋白去乙酰化酶的活性,增加组蛋白H3赖氨酸9的乙酰化,从而提高参与脂肪生成和脂肪酸摄取的基因表达[18]。
糖酵解活性的增强可促进线粒体ROS的产生,过量的ROS将攻击细胞内大分子化合物,如蛋白质、核酸和脂质等,并导致细胞死亡或诱导细胞凋亡,这也是NAFLD进展为NASH的重要因素之一。线粒体是细胞内ROS的重要来源,线粒体ROS(mtROS)主要在线粒体内膜中电子传递链的氧化磷酸化过程中产生,当mtROS增加时,由于线粒体功能障碍,诸如线粒体DNA(mtDNA)之类的线粒体损伤相关分子模式被释放至细胞中或细胞外,mtROS被氧化以促进mtDNA的细胞质转运,氧化后的mtDNA直接与含有核苷酸结合结构域-、富含亮氨酸重复序列-和NOD样受体热蛋白结构域相关蛋白3结合,刺激IL-1β的产生,加速NAFLD进展[19]。因此,通过抑制mtROS,维持线粒体稳态,或可作为NAFLD的潜在治疗手段。一项关于巨噬细胞和NASH的研究[20]发现,膜联蛋白A5通过直接靶向PKM2促进活性PKM2四聚体的形成,导致糖酵解抑制和线粒体氧化代谢激活,从而触发巨噬细胞表型转移和改善NASH,为NASH提供了一种新的治疗方法。
3.2 肝纤维化 广泛的慢性肝损伤,包括病毒性肝炎、胆汁淤积性肝病和NAFLD,可引起慢性肝脏炎症并最终导致肝纤维化。如果能够去除致病因子,肝纤维化是可逆的[21]。而当无法去除潜在病因时,早期识别、预防肝纤维化则成为临床治疗的关键。肝纤维化的特征在于肝星状细胞(HSC)的激活、增殖和迁移。活化的HSC进一步促进过量胶原蛋白的形成和细胞外基质(ECM)的积累,导致持续性慢性肝损伤,若不及时加以干预,最终将进展为肝硬化和肝细胞癌。肝脏作为人体最大的代谢中心,探寻肝纤维化相关的代谢途径改变可能有望发现肝纤维化的新标志物和治疗靶点。研究[22]显示,HSC在活化过程中存在有氧糖酵解,抑制有氧糖酵解中的LDH-A可阻断HSC收缩。另有研究[23]发现,葡萄糖转运蛋白(glucose transporter,GLUT)1和PKM2在临床患者和小鼠纤维化肝脏样本中的表达均上调,且来自活化的 HSC外泌体中 GLUT1 和 PKM2表达显著增加,表明HSC释放的外泌体与HSC的活化和葡萄糖摄取相关,并通过运输糖酵解相关蛋白进而影响肝脏非实质细胞(包括静止型HSC、Kupffer细胞和肝窦内皮细胞)向糖酵解的代谢转换。最近一项研究[24]发现,局灶性黏附激酶相关非激酶(FAK related non-kinase,FRNK)通过抑制有氧糖酵解,限制HSC的活化、增殖和迁移,并促进HSC凋亡,从而改善肝纤维化,表明FRNK可能是肝纤维化治疗的潜在治疗候选药物。Rao等[25]利用特异性卵泡抑素样蛋白1(follistatin like protein 1,FSTL1)基因敲除小鼠构建肝纤维化模型以探索巨噬细胞FSTL1在肝纤维化中的功能和机制,结果发现,FSTL1促进了PKM2磷酸化和核易位,减少了PKM2泛素化以增强PKM2依赖的糖酵解,并增加了M1极化,在使用PKM2活化剂(DASA-58)后,FSTL1介导的糖酵解和炎症表现受到部分抑制。关于PKM2在肝纤维化中的作用已被证实,PKM2四聚化可逆转肝纤维化,诱导PKM2四聚化以降低PKM2二聚体的水平可能是肝纤维化的潜在治疗策略[26]。Zhou等[27]在肝纤维化小鼠肝脏中发现转化生长因子β1(TGFβ1)可通过激活Smad、p38丝裂原活化蛋白激酶和P13K/AKT信号通路,引起HSC中有氧糖酵解的增加,并诱导HSC中的GLUT1表达,促进肝纤维化的发展,而在肝纤维化的小鼠模型中使用GLUT1抑制剂后,肝脏炎症和肝纤维化的程度明显降低。Ban等[28]研究发现,木香烃内酯可以抑制HK2的表达,进而抑制HSC活化,表明木香烃内酯抑制糖酵解可能是治疗肝纤维化的潜在策略。HSC激活是肝纤维化过程中的核心环节,有氧糖酵解是其代谢标志之一。因此,阻断糖酵解或将成为肝纤维化的一种新型治疗选择。
3.3 肝细胞癌 早在1956年,德国生理学家奥托·海因里希·瓦尔堡(Otto Heinrich Warburg)发现癌细胞更喜欢通过糖酵解消耗大量葡萄糖,而不是倾向于利用氧化磷酸化,即使在足够氧气的条件下也倾向于将葡萄糖转化为乳酸盐,这种现象被称为有氧糖酵解或“Warburg效应”。尽管在有氧糖酵解过程中ATP的产生效率较低,但在不同肿瘤中仍占ATP供应量的50%~70%[29]。此外,在有氧糖酵解过程中产生的中间体可用于肿瘤生物大分子的生物合成,以满足快速生长的需求,乳酸的产生也提供了一个酸性环境,以帮助癌细胞的侵袭和转移[30]。HCC中有氧糖酵解的分子机制是一个复杂的过程,既包含细胞质中糖酵解酶活性的改变,也包含细胞核中的遗传变化,靶向抑制有氧糖酵解中关键酶的变化将在很大程度上影响HCC的进展。
HK是有氧糖酵解中的第一种限速酶,可催化葡萄糖转化为葡萄糖-6-磷酸(G-6-P)。HK有4种亚型,即HK1、HK2、HK3和HK4,但大多数正常组织仅表达HK1。然而,HK2在HCC组织中高度表达[31],并且相对于其他亚型,HK2在促进有氧糖酵解方面更有效。首先,HK2与线粒体外膜的电压依赖性阴离子通道蛋白1(voltage-dependentanionchannels,VDAC1)相互作用并结合,促进ATP合成相关酶的活化,以增强ATP的生成,并抑制细胞凋亡(图1)。其次,当HK2与VDAC1结合时,其不会受到下游产物如G-6-P的抑制作用,从而增强糖酵解过程和提高ATP生成速率[32]。HK2的消耗可抑制糖酵解通量并诱导氧化磷酸化,增强HCC对药物(如二甲双胍)的敏感性,此外,HK2抑制剂还可协同增强HCC对索拉非尼的敏感性,从而抑制小鼠的肿瘤生长[31]。因此,基于HK在HCC中的关键作用,HK2或将成为开发HCC新疗法的靶点。
磷酸果糖激酶1(phosphofructokinase 1,PFK1)是参与糖酵解的第2种限速酶,可利用ATP催化果糖6-磷酸(F-6-P)转化为果糖-1,6-二磷酸(FDP)。PFK1有3种亚型,分别为PFK-M、PFK-P和PFK-L,这些亚型的比例在不同组织中可能根据其特定的能量代谢需求而有所不同[33]。完全活化的PFK1以四聚体的形式存在,PFK1四聚体的形成和稳定在很大程度上影响糖酵解通量速率。6-磷酸果糖-2-激酶/果糖-2,6-双磷酸酶3(PFKFB3)分子是PFK1的变构激活剂,可催化果糖-2,6-二磷酸(F-2,6-BP)的产生。PFKFB3在HCC糖酵解的调节以及肿瘤生长和转移中发挥重要作用,PFKFB3可易位至细胞核中,以调节细胞周期依赖性激酶(cyclindependent kinase,CDK)的活性,导致细胞周期阻滞并抑制细胞死亡[34](图1)。研究[35]发现,在抑制PFKFB3的表达后联合使用阿司匹林与索拉非尼,可通过诱导HCC细胞凋亡来克服索拉非尼的耐药性,以增强HCC的治疗效果。因此,在寻找化疗药物与糖酵解抑制剂的组合时,运用PFKFB3抑制剂可能成为克服索拉非尼耐药性的有效手段。
糖酵解过程中的最后一种限速酶是丙酮酸激酶(PK),其催化磷酸烯醇式丙酮酸产生ATP和丙酮酸。PK具有4种亚型,包括PKL、PKR、PKM1和PKM2,其中PKM2在癌细胞中高度上调,并且与预后不良有关[36]。PKM2有2种形式,一种是位于细胞质中的四聚体,具有较高的催化活性,可以迅速将磷酸烯醇式丙酮酸转化为丙酮酸、增加糖酵解通量和产生更多的ATP;另一种亚型是单体或二聚体,具有较低的催化活性,并且可以易位至细胞核中作为几种转录因子[例如缺氧诱导因子-1α(hypoxia inducible factor-1,HIF-1α)、β-catenin/c-Myc、核因子κB(nuclear factor kappa-B,NF-κB)、信号转导和转录激活因子3(signal transducer and activator of transcription 3,STAT3)]的共激活剂[37](图1)。一旦进入细胞核,PKM2可促进靶基因的转录,例如HIF-1α靶向表达GLUT、PKM2、LDH-A和血管内皮生长因子A,从而促进癌细胞的生长,正反馈调节糖酵解和血管生成[38]。PKM2的表达水平与HCC的临床病理特征有关,例如肿瘤的大小、数量和临床分期,并且PKM2表达水平较高的HCC患者相较于PKM2水平较低的HCC患者表现出更高的复发率[39]。紫草素(shikonin)和原花青素B2可抑制PKM2的表达和有氧糖酵解的发生,从而抑制HCC的生长,表明PKM2有望成为治疗HCC的又一治疗靶点[40-41]。
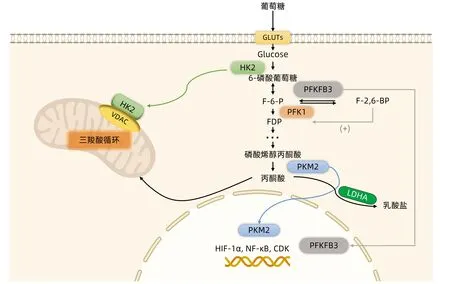
注:HK2、PFK1和PKM2是糖酵解过程中的3种限速酶。HK2可催化葡萄糖生成6-磷酸葡萄糖,并与线粒体外膜上的VDAC1相互作用和结合,从而促进ATP的产生和抑制细胞凋亡。PFK1可催化F-6-P为FDP,其活性受PFKFB3催化产物F-2,6-BP的调节。PKM2不仅能够催化PEP合成丙酮酸,还能够移位至细胞核内,与一些转录因子如HIF-1α、β-catenin/c-Myc、NF-κB和STAT3共同激活,促进相关靶基因的转录。
4 小结与展望
糖酵解与线粒体功能障碍密切相关,由异常糖酵解引起的线粒体功能障碍与ROS产生轴之间的恶性循环促使肝病进一步恶化。在NAFLD肝细胞中,糖酵解显著增强,导致血浆和肝脏中丙酮酸盐的水平增加。丙酮酸也可以通过为三羧酸提供中间体转化为草酰乙酸或乳酸,两者在NAFLD中均增强。糖酵解活性的增强将促进线粒体ROS的产生,导致NAFLD进展为NASH,抑制mtROS维持线粒体稳态或可作为NAFLD的潜在治疗方案,阻止疾病进一步发展。越来越多的证据表明,HSC的活化过程中存在有氧糖酵解,在肝纤维化中,部分相关代谢途径发生改变,因此通过一些代谢途径阻断糖酵解或将成为肝纤维化的一种新型治疗选择。有氧糖酵解在HCC的进展中发挥重要作用,包括增殖、免疫逃避、侵袭转移、血管生成和耐药性,通过靶向有氧糖酵解中包含的关键酶(例如HK2、PFK或PKM2)以及其他调节途径对于突破当前HCC治疗中的局限性可能具有重要意义。反观肝衰竭,其核心机制为内毒素、免疫反应和炎症级联反应等[42],并且糖酵解参与其中[43]。笔者所在团队前期研究[44]发现,解毒化瘀颗粒可通过调节急性肝衰竭大鼠肝线粒体PT孔开放,阻止线粒体通透性转换,抑制细胞色素C的释放,影响caspase-3蛋白活性,从而抑制肝细胞凋亡。同时,还可能通过促进PI3K/AKT/mTOR信号通路表达进而有效改善肝细胞氧化应激与线粒体受损[45]。在往后的研究中,可以尝试以糖酵解与线粒体功能障碍之间的关系为落脚点,探索两者在肝衰竭疾病进展过程中的代谢途径变化,开展对肝衰竭进展的把控以及治疗靶点的研究。
利益冲突声明:所有作者均声明不存在利益冲突。
作者贡献声明:颜耿杰负责课题设计,资料分析,撰写论文;林镛、苏会吉、陈含笑、班少群参与收集数据,修改论文;毛德文、韦艾凌、龙富立负责拟定写作思路,指导撰写文章并最后定稿。
