乙型肝炎病毒在弥漫性大B细胞淋巴瘤发生发展中的作用
2022-09-07陈宏森何奕达李子帅王瑞华曹广文
陈宏森 何奕达 李子帅 王瑞华 曹广文
作者单位:200433 上海 1第二军医大学海医系流行病学教研室;510632 广州 2暨南大学流行病学教研室
全球大约有2.96亿人感染乙型肝炎病毒(hepatitis B virus,HBV)[1],其中我国HBV感染人数为9 400万左右[2]。HBV是导致肝细胞癌(hepatocellular carcinoma,HCC)的主要致病因素[3]。然而,流行病学研究发现HBV除了与HCC相关外,还与非霍奇金淋巴瘤(non-Hodgkin′s lymphoma,NHL)相关[4]。NHL中最常见的侵袭性肿瘤为弥漫性大B细胞淋巴瘤(diffuse large B cell lymphoma,DLBCL),占NHL的30%~40%。近年来,不管是全球的NHL发病率还是我国的NHL发病率均呈上升趋势[5-7]。因此,探索NHL的病因也成为研究者们关注的课题。本文就HBV导致DLBCL的发病机制进行综述,以期为其临床诊疗提供新的思路。
1 HBV感染是导致DLBCL的病因之一
DLBCL包括活化B细胞样(activated-B cell like,ABC)亚型和生发中心B细胞(germinal center B-cell,GCB)亚型,致病因素主要包括遗传因素、免疫因素、职业暴露、感染等。其中EB病毒(epstein-barr virus,EBV)感染、丙型肝炎病毒(hepatitis C virus,HCV)感染、人类免疫缺陷病毒(human immunodeficiency virus,HIV)感染等都会导致DLBCL的发生[4]。结合世界各国(地区)多项研究发现,HBV感染也可导致DLBCL,且HBV感染相关DLBCL具有如下临床特点[8-13]:HBsAg阳性率高(约20%),脾脏累及更加明显,诊断年龄更年轻(中位年龄约为42岁),国际预后指数高(预后差,尤其是GCB亚型),诊断时疾病分级多为晚期(通常为Ⅲ/Ⅳ期),治疗上具有化疗抵抗等特点。目前已有多项研究显示HBV感染会增加DLBCL 发病风险[8,13-20]。在一项关于美国66岁以上老年人群的病例对照研究中发现,HBV感染不仅会增加肝癌的发生风险(OR=10.60,95%CI:9.66~11.60),也会增加DLBCL的发生风险(OR=1.24,95%CI:1.06~1.46),但与T细胞型NHL、其他B细胞型NHL无关[14]。LI等[16]也报道HBsAg阳性是B细胞型NHL的危险因素(OR=3.26,95%CI:2.33~4.84),但未发现其是T细胞型NHL的危险因素(OR=2.13,95%CI:0.92~4.92)。我国和新加坡3项前瞻性队列的汇总研究也表明,HBsAg阳性可以显著增加NHL的发病风险(OR=1.88,95%CI:1.61~2.20)[17]。台湾地区的队列研究显示,HBV感染者DLBCL发病风险较HBV阴性者提高了约1.69倍(aHR=2.69,95%CI:2.05~3.52)[18],在另一项台湾地区的回顾性队列研究中HBV(HR=2.49,95%CI:1.94~3.19)和 HCV(HR=2.36,95%CI:1.73~3.22)感染患者的NHL发病率也显著高于普通人群,表明HBV和HCV感染显著增加了NHL的风险[20]。同样的,基于我国人群的队列研究也显示,HBsAg血清反应阳性的参与者(n=15 355)与HBsAg血清反应阴性参与者(n=481 377)相比,患淋巴瘤的风险提高了2.10 倍(HR=2.10,95%CI:1.34~3.31)[19]。一项 Meta分析汇总了国内外HBsAg与NHL相关性的研究结果[15],同样发现HBsAg阳性是NHL的危险因素(aOR=1.95,95%CI:1.55~2.44,I2=90.3%),其中我国NHL患者(PooledOR=2.40,95%CI:1.56~3.68,I2=95.2%)相比非我国患者(PooledOR=1.81,95%CI:1.34~2.45,I2=88.9%)以及血清HBsAg阳性患者罹患NHL的可能性更高,说明HBV血清标志物在DLBCL致病过程中可能发挥一定作用。
目前,临床试验研究也表明HBV感染与DLBCL预后密切相关,且预防性抗病毒治疗可以改善预后。YAMAUCHI等一项关于含利妥昔单抗化疗的HBsAg阳性DLBCL患者的预防性抗病毒治疗研究,在HB-sAg阳性患者中比较了接受恩替卡韦或拉米夫定与未接受抗病毒治疗患者的生存情况,发现未接受抗病毒治疗的HBsAg阳性DLBCL患者4年无进展生存率较差(44.4%,95%CI:13.6%~71.9%),而接受拉米夫定治疗患者(79.7%,95%CI:54.5%~91.9%)或恩替卡韦治疗患者(66.0%,95%CI:54.6%~75.2%)的4年无进展生存率相对较好(P=0.047)[21]。一项基于人群的研究也证实了预防性抗病毒治疗对HBsAg阳性DLBCL患者的重要性,其中HBV感染后未进行抗病毒治疗的DLBCL患者中位生存期明显较接受抗病毒治疗的DLBCL患者缩短(35.61个月vs74.23个月,P=0.003),且HBV感染DLBCL患者抗病毒治疗后与HBV阴性患者的中位生存期相当(分别为74.23个月以及未达到,P=0.002)[22]。
总体来看,HBV感染与B细胞型NHL(尤其是DLBCL)显著相关,HBV感染是导致NHL(尤其是DLBCL)的病因之一,而非肿瘤发生后免疫功能下降引起HBV感染。但是HBV可能是DLBCL的一个必要但不充分因素,主要通过与其他致病因素(如感染、职业暴露、免疫抑制等)协同作用,导致DLBCL的发生。然而,DLBCL具有高度异质性,分子亚型众多[23],其致病因素仍需进一步研究阐明。
2 HBV致DLBCL的可能机制
关于HBV感染在DLBCL发生发展中的可能机制,现有研究主要根据三种理论开展,即慢性抗原刺激理论、HBV直接感染淋巴细胞理论及隐匿性HBV感染相关理论。
2.1 慢性抗原刺激理论
慢性抗原刺激理论依据HCV感染导致DLBCL提出[24-25],该理论认为受HBV抗原慢性刺激的B细胞发生恶性转化,其中恶性转化包括双链DNA断裂、染色体易位、原癌基因过表达等突变事件的发生。HCV包膜蛋白E2(envelope protein 2,E2)是人体B细胞抗HCV的主要靶点,HCV-E2与B细胞表面的CD81结合,刺激B细胞增生,随后多克隆B细胞持续激活,从而促进B细胞恶变[26]。慢性抗原刺激理论的具体作用机制见图1。

图1 慢性抗原刺激理论的作用机制Fig.1 The mechanism of chronic antigen stimulation hypothesis
2.1.1 HBsAg特异性B细胞 慢性乙型肝炎(chronic hepatitis B,CHB)患者的血液和肝脏中可提取到HBsAg特异性B细胞(HBsAg-specific B cell),这种B细胞能持续存在于患者的肝脏和血液中,且因功能缺陷无法产生有效的抗体。不仅是HBsAg特异性B细胞受损,患者其他的B细胞功能也会受损,这使机体对病毒产生免疫耐受或免疫耗竭[27]。基因表达分析发现,B细胞抗原刺激相关的基因(如CD83、NFKBIZ等)均呈现上调特征。其中NFKBIZ基因突变与NF-κB通路异常及促癌基因失调相关,这可导致ABC亚型DLBCL的发生[28]。与HCV感染、HIV感染患者血细胞研究结果一致,从CHB患者提取的B细胞中还有大量非典型记忆B细胞(atypical memory B cells,atMBC),这些细胞表现出信号通路改变、归巢、产生抗病毒以及促炎细胞因子(CD11c和CXCR3)的特征,且可高表达抑制性受体(如PD-1等),一方面影响机体抗病毒能力,一方面可使异常B细胞产生免疫逃逸表型[29-30]。
2.1.2 HBcAg特异性B细胞 与HBsAg相比,外周血循环中的B细胞更容易对HBcAg产生免疫效应,且HBcAg特异性B细胞较HBsAg特异性B细胞数量更多、抗体反应更强。HBsAg和HBcAg特异性B细胞具有与整体记忆细胞不同的mRNA表达模式,除产生抗体外,还有交叉呈递、固有免疫相关基因高表达的特征[31-32]。机体感染HCV后,也会产生atMBC,这种B细胞可产生有类风湿因子活性的抗体,这种抗体能够结合人类自身IgG Fc区的独特表位且持久存在,at-MBC产生的抗体结合IgG Fc区表位与HCV慢性感染导致的自身免疫病或淋巴瘤相关[33]。慢性抗原刺激导致HBV感染的过程可能是B细胞持续暴露于HBV抗原而产生了功能受损的atMBC,atMBC周期性激活,在接受外来抗原刺激后会重新进入生发中心,进入生发中心的atMBC反复暴露于AID介导的异常体细胞高频突变(somatic hypermutation,SHM),使错义突变积累,导致B细胞克隆增殖和表型改变,最终使记忆B细胞发生恶性转化和免疫逃逸[34]。
2.1.3IGVH基因片段偏好性使用 DLBCL能以BCR的形式表达免疫球蛋白重链可变区(immunoglobulin variable heavy chain region,IGVH)基因,不仅可作为克隆标志物,还可为恶性B细胞提供线索来源。HBV感染的DLBCL患者IGVH基因片段在Ig基因重排时具有偏好性使用特点,即不同患者可表达几乎相同的BCR基因片段序列[35],这可以作为支持慢性抗原刺激理论最有力的证据。HBV在DLBCL中的高流行率,尤其是HBsAg的高阳性率暗示HBsAg在肿瘤生成中可能发挥重要作用,而这可能与慢性抗原持续刺激相关。然而,基因测序比对发现[8],HBsAg阳性DLBCL患者IGVH基因没有偏向性使用(biased usage),且HBsAg阳性DLBCL患者CDR3区域没有固定型,与乙肝病毒表面抗体(anti-HBs)也不存在任何相同序列。总之,目前慢性抗原刺激理论的相关证据和文献还较少,且研究结论尚未统一,因此仍需进一步研究验证。在HBV感染中,HBsAg和HBcAg特异性的B细胞在DLBCL致病过程中的具体作用,是否与HCV相关DLBCL致病机制一致也尚未有定论。但是总体上目前更多的研究结果支持HBV感染淋巴细胞导致DLBCL这一观点。
2.2 HBV直接感染淋巴细胞致淋巴瘤
HBV除了具有嗜肝性外,还能感染淋巴细胞。HBV进入人体后,B细胞能将病毒颗粒、HBsAg“内化”[36],之后HBV可以随B细胞进入淋巴系统,且在淋巴结这一“贮存池”中长期存在。HBV感染B细胞后主要起两方面致癌作用:⑴人体中载脂蛋白B mRNA编辑酶催化多肽(apolipoprotein B mRNA-editing enzyme catalytic polypeptide-like,APOBEC)家族发挥效应,介导异常体细胞高频突变,产生淋巴瘤;⑵HBV基因组整合到宿主基因,驱动致癌突变。HBV直接感染淋巴细胞致淋巴瘤的具体机制见图2。

图2 HBV直接感染淋巴细胞导致淋巴瘤的机制Fig.2 The mechanism of HBV direct infection lymphocytes leading to lymphoma
APOBEC家族是人体内具有抗病毒活性的酶,在肝癌、肺癌、胃癌、肾癌等多种肿瘤生成中起重要作用。其中,APOBEC3A和胞苷脱氨酶(activation induced cytidine deaminase,AID)可引发病毒基因或免疫球蛋白发生高频突变。最近,多种癌症集群突变图谱发现,高达76.1%的“突变雨”(kataegis,基因组中一种长链协调性突变)、16.2%的“突变雾”(omikli,基因组中一种弥漫性突变)与AID和APOBEC3家族相关[37-38]。APOBEC家族在DLBCL中(无论伴或不伴HBV感染)扮演了重要角色,而AID与SHM、免疫球蛋白类别转换重组(class switch recombination,CSR)有关,SHM、CSR这两个过程异常在DLBCL生成中起关键作用[39]。在关于DLBCL分子亚型和致病机制的研究中发现[40],DLBCL的突变标签,80%为“衰老”标签,该标签主要为CpG位点的自发脱氨,与人体衰老引发的自发脱氨突变有关(DLBCL主要病因为衰老)。此外,c-AID和AID2这两个AID驱动的突变标签反映了AID诱导胞嘧啶脱氨基成为尿嘧啶(C>U)后两种不同的修复机制。其中,c-AID的特征是在已知的AID“热点”区[RCY-motif(R=A/G,Y=C/T)]C>T/G突变增加,且c-AID在生理性和异常体细胞高频突变部位较活跃;AID2的特征是以(WA[W=A/T]-motifs)位置上A>T/C/G突变为主。
通过采用全基因组测序/全外显子组测序(WGS/WES)、转录组测序、靶向测序等技术分析发现,HBV感染的DLBCL基因突变明显增强,且具有独特的突变图和特征[8]。整合WGS/WES和淋巴芯片数据,发 现 有 14个 基因 突变(KLF2、TMSB4X、CD70、BCL6、FAS、TNFRSF14、UBE2A、CD58、SGK1、ZFP36L1、CXCR4、FOXO1、CSK和MSL2)偏向于发生在HBsAg阳性DLBCL患者中,其中KLF2、TMSB4X、CD70、BCL6、FAS、TNFRSF14、SGK1、ZFP36L1、CXCR4、FOXO1、CSK这11个为活化诱导AID潜在的脱靶基因。突变标签分析发现了与APOBEC相关的标签,说明HBV感染相关DLBCL患者基因突变频率增加,且部分为APOBEC/AID所致。B细胞由于慢性感染高度活化,经AID介导的基因突变增强,进而诱导反复发生的体细胞突变或结构变异,最终形成在HBV感染的DLBCL人群中观察到的突变特征。HBV感染相关DLBCL患者中最关键的突变通路与HBV感染相关通路涉及的基因突变一致,这些基因包括MYD88、GRB2、BCL2、TP53、NFKBIA、FAS、MYC、STAT3、PRKCB。此外,在HBV感染相关DLBCL患者中发现的高频基因突变还涉及其他重要信号通路,包括p53、FOXO、BCR、JAK-STAT、NF-κB以及表观遗传修饰、免疫逃逸和细胞迁移等相关信号通路。有研究通过单细胞RNA测序(scRNA-seq)分析发现HBV对ABC亚型和GBC亚型肿瘤细胞的生存及免疫逃逸均有重要影响[41],其中HBV感染相关DLBCL中GCBC亚型的主要组织相容性复合体Ⅱ(major histocompatibility complex Ⅱ,MHC Ⅱ)分子表达缺失并促进了肿瘤细胞逃逸。而ABC亚型的肿瘤细胞虽然高表达MHCⅡ分子,但会招募更多具有潜在免疫抑制功能的Treg细胞,且可以通过CD40LG-CD40轴从CD4+T细胞接收更多的反向促增殖信号,进而促进肿瘤细胞的免疫逃逸和生长。另外,HBV感染相关DLBCL的微环境中显著富集了一组记忆性B细胞,这些细胞高表达HBx靶基因EGR2和EGR3,且与前文提到的atMBC在功能上相似。
HBV感染淋巴细胞后,病毒基因还会在宿主基因组内整合。在HBV相关HCC中,有80%~90%的患者可以观察到HBV整合,整合后主要通过两种机制参与HCC发展,即顺式激活作用和反式激活作用。TERT、MLL4和CCNE1基因已被反复确认为HBV在HCC中的优先整合基因位点[42]。大多数情况下,HCC中整合的HBV DNA包含病毒基本核心启动子(BCP)/增强子Ⅱ(Enh Ⅱ)和编码C端截短型HBx基因(C-terminal truncated HBx,ct-HBx)。其中,ct-HBx是一种多效性反式激活因子,在HBV整合过程中发挥重要作用,且HCC患者ct-HBx表达量较HBx显著升高[16,22]。
与HBV相关HCC类似,在大约50%的HBV相关NHL肿瘤组织中也能鉴定出HBV整合的DNA[16],但是NHL中的HBV DNA整合位点与HCC不同。结合高通量病毒整合检测(high-throughput viral integration detection,HIVID)和免疫组化分析发现,HBV DNA在淋巴瘤组织中可反复靶向整合7个蛋白质编码基因,即ANKS1B、CAPZB、CTNNA3、EGFLAM、FHOD3、HDAC4和OPCML,这些基因在NHL中的表达水平显著改变,可能是DLBCL的潜在候选致癌基因。6个非编码RNA 基 因,即LINC00499、LINC00603、LINC01360、LINC00486、MIR7976和PWRN1也被HBV DNA反复靶向整合。HBV整合片段主要在S基因的3′端,位于nt.273-465内。从NHL分离的整合preS/S序列编码MSt蛋白(C-terminal truncated middle S protein),其C端有123~187个氨基酸的截短,这种MSt蛋白具有转录活性及反式激活功能。MSt反式激活子的持续表达可能有利于病毒在宿主细胞中生长,而DLBCL的发生、肿瘤细胞的持续存在可能与其相关。
2.3 隐匿性HBV感染与病毒准种
HBsAg阴性人群罹患DLBCL的病因与隐匿性HBV感染和HBV准种有关。隐匿性HBV感染指血清HBsAg检测为阴性,但肝脏中存在具有活跃复制能力的HBV DNA,血液中有(或无)HBV DNA存在,既往有(或无)HBV感染史。HBV进入宿主细胞后,部分双链环状DNA进入细胞核,形成HBV共价闭合环状DNA(covalent close circle DNA,cccDNA)。隐匿性HBV感染与肝脏中HBV共价cccDNA持久稳定存在有关。现有治疗手段无法有效清除cccDNA,HBV感染者一生都可存在cccDNA。cccDNA是HBV潜伏期主要的具有转录活性的病毒模板,在细胞核中以游离状态存在。利用巢式PCR、二代测序及分子克隆技术可检测DLBCL患者中的隐匿性HBV感染、cccDNA和病毒准种情况[43],从而了解隐匿性HBV感染导致DLBCL的机制。病毒准种是指一大群在遗传学上相关但基因不完全相同的HBV变体。由于HBV高复制且易出错的倾向,HBV变体在宿主内积累,并在免疫系统筛选下持续地遗传变异、竞争和选择,特异性的变异株被筛选出来,且在人体合适生态位上成为优势准种并长期潜伏[44]。由于血液中免疫监视下的选择压力更大,相较肿瘤组织或癌旁组织,HBV在血液中的进化速度更快。因此,HBV准种复杂性更低,也更容易出现优势准种[45]。HBV准种长期存在,使血液中的B细胞慢性激活,在其他致癌因子共同作用下,具有潜在致癌性的准种驱动DLBCL形成,隐匿性HBV感染与病毒准种导致DLBCL的机制如图3所示。
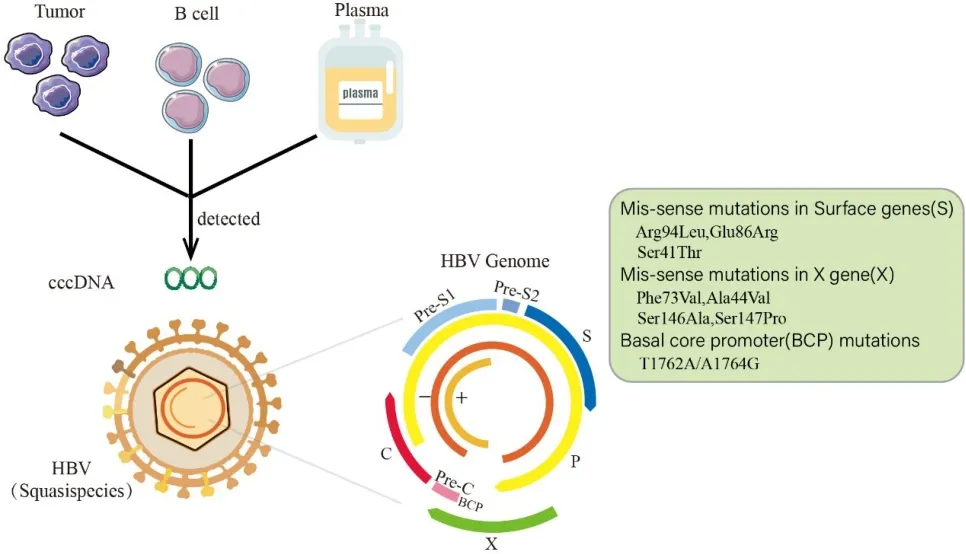
图3 隐匿性HBV感染与病毒准种导致DLBCL的机制Fig.3 The mechanism of occult HBV infection and viral quasispecies leading to DLBCL
有研究[46]在DLBCL患者中检测发现约67.5%的患者为隐匿性HBV感染患者,且这些患者中约41%可在血浆、B细胞或福尔马林固定、石蜡包埋样本中检测到cccDNA;通过二代测序分析发现,在血浆、B细胞、肿瘤组织区室之间,发生在核心(C)基因的错义突变有B细胞区的Arg94Leu、Glu86Arg和DLBCL肿瘤区的Ser41Thr,且Ser41Thr特异性存在于肿瘤组织上,因此可能是DLBCL的候选突变基因。X基因的错义突变有反式激活区(Phe73Val)、X结合区(Ala44Val)和p53结合区(Ser146Ala,Ser147Pro)。其中,X结合区(Ala44Val)在上述研究中被认为是HCC的致病因素(OR=7.6,95%CI:1.4~53.7),可能在DLBCL致病过程中起作用[46]。此外,该研究还在肿瘤组织内发现了基本核心启动子区的经典双突变T1762A/A1764G,而该突变不仅与HCC密切相关,也可能在DLBCL发生中起作用[46]。我国是乙肝大国,但乙肝诊断率仅为25%[47],因此预计隐匿性HBV感染人口数量较多。Meta分析显示,全球人口的BQA感染率在0%~35.61%波动,总体隐匿性HBV感染流行率为0.82%[48]。由于隐匿性HBV感染患者有罹患DLBCL的潜在风险,因此在输血、器官移植等临床诊疗过程中,还需加强隐匿性HBV感染筛检,以避免隐匿性HBV感染的社区传播。
3 HBV在DLBCL研究及治疗中的意义
HBV导致B细胞的恶变符合“变异-选择-适应”理论,即HBV慢性感染B细胞后在细胞因子的作用下,诱发并维持慢性非可控性炎症,APOBEC家族介导致癌基因突变,激活相关通路。同时,HBV cccDNA可长期存在于人体中,使HBV整合进入人体基因组,从而导致HBV基因在宿主体内长期进化。经过宿主免疫系统筛选出HBV准种,在HBV准种长期刺激下,B细胞发生驱动致癌突变,这符合“癌症进化发育学”学说和宿主基因组选择变异规律。
目前,美罗华(利妥昔单抗)已成为DLBCL治疗不可或缺的成分,美罗华联合CHOP化疗方案大大提高了DLBCL患者的生存率[49-50]。然而,临床治疗中也存在HBV感染DLBCL患者出现化疗抵抗、肝功能受损甚至死亡事件[10,51],因此,研究 HBV 感染致DLBCL的发病机制、隐匿性HBV感染与DLBCL关系的研究等对临床治疗也有着重要意义。有研究[9]发现,在HBV感染DLBCL患者中,lncNBAT1对患者化疗效果有积极影响。该研究发现HBV的HBx可上调lncNBAT1表达,而lncNBAT1与信号转导子和转录激活子 1(signal transducer and activator of transcription,STAT1)相互作用可以阻止STAT1在APOBEC3A的启动子区域富集;HBx/lncNBAT1还可抑制APOBEC3A的表达,降低化疗药物阻滞DLBCL细胞S期的作用效果,从而产生化疗抵抗作用。因此,lncNBAT1可能是HBV感染DLBCL患者的候选治疗靶点。APOBEC3A则可能是潜在的预后标志物[9]。此外,单细胞RNA测序发现[41],DLBCL中的T细胞免疫球蛋白和免疫受体酪氨酸抑制性基序结构域(T cell immunoreceptor with Ig and ITIM domains,TIGIT)以及甲型肝炎病毒细胞受体2是T细胞耗竭和失能的主要因素。因此,针对共抑制信号TIM3或TIGIT,再联合PD-1信号通路开展联合免疫治疗较单独针对PD-1的免疫治疗疗效可能更好。虽然该发现主要与甲型肝炎病毒感染DLBCL相关,但对HBV感染相关DLBCL的治疗也有重要意义,未来若能发现HBV相关DLBCL细胞抑制性免疫受体,针对该分子开展综合免疫治疗也具有可行性。
4 小结与展望
DLBCL具有异质性,分类、分型方法较多,致病机制至今尚未完全清楚。近年来研究发现HBV感染与DLBCL密切相关,可能是DLBCL的致病因素之一,且认为HBV在DLBCL发生、发展中的可能作用包括以下三个方面:⑴HBV抗原在人体外周血中可以作为致病抗原长期反复刺激记忆B细胞,由病毒刺激产生的B细胞功能受损,无法产生有效抗体,但可以产生细胞因子和炎症因子。功能受损B细胞再次进入生发中心,反复暴露于APOBEC/AID家族介导的SHM和CSR,这一过程称为慢性抗原刺激。⑵HBV可直接感染淋巴细胞,病毒颗粒及抗原被B细胞“内化”,病毒基因组在细胞中整合,致使细胞致癌突变增强,最终驱动DLBCL发生。⑶在隐匿性HBV感染人群中鉴定到的HBV基因突变,说明HBV还可以通过隐匿性感染长期存在人体内,病毒准种进化形成优势准种,驱动B细胞发生致癌突变。如前文所述,目前HBV相关DLBCL的研究已经取得了一些进展,但是这些研究总体上样本量均较小、差异表达分子也有待进一步挖掘、分子层面的致病机制研究也不够深入,因此HBV致DLBCL的发病机制仍需进一步深入探索。此外,由于我国的HBV诊断率和检出率不高,但存量HBV感染患者多且存在众多隐匿性感染人口,加之近年来NHL发病率呈上升趋势,因此开展HBV感染相关DLBCL研究尤为必要,这不仅可以阐明HBV感染在DLBCL发生发展中的具体致病机制,也有助于提高DLBCL治疗的特异性和靶向性。
