肠道微生物测序研究中不同实验来源差异的综合评价以及应用
2022-09-07徐珂琳朱嗣博薛江莉蒋艳峰袁子宇王久存张铁军陈兴栋
徐珂琳, 庄 悦, 朱嗣博, 薛江莉, 蒋艳峰, 袁子宇,王久存, 索 晨,5), 张铁军,5), 吕 明,6), 陈兴栋*
(1)复旦大学公共卫生学院生物统计学教研室, 上海 200032;2)复旦大学泰州健康科学研究院,江苏, 泰州 225316;3)复旦大学生命科学学院现代人类学教育部重点实验室, 上海 200433;4)复旦大学人类表型研究所, 上海 201203;5)复旦大学公共卫生学院流行病学教研室, 上海 200032;6)山东大学齐鲁医院临床流行病学研究中心, 济南 250012)
人类胃肠道具有大量和高度多样化的微生物群,是至今研究最多的生态系统之一。由于肠道微生物群在人体健康和内稳态方面的重要性,人们对其进行了详细的研究。代谢紊乱[1-4]、炎症性肠病[5, 6]和癌症[7]等疾病的研究已经揭示了肠道中特定的生物标志物和生态系统。大规模的研究,例如人类微生物组计划(Human Microbiome Project, HMP)和人类肠道宏基因组学(Metagenomics of the Human Intestinal Tract, MetaHIT),为进一步的宏基因组学研究提供了必要的参考[8, 9]。这些研究采用16S rRNA扩增子测序(16S rRNA基因测序或16S测序)或宏基因组鸟枪(whole-metagenome shotgun, WMS)测序方法进行分类学图谱分析。
近年来,WMS法被应用于许多微生物组图谱研究,在微生物组成和代谢通路的解释方面展现出巨大的潜力[8, 10-13]。在WMS中,确定宏基因组分析和数据分析中的技术和生物学变异至关重要。此外,使用不同测序仪和文库制备方法的肠道微生物测序研究中的分类学图谱通常不一致,合并这类研究的结果仍然是一个亟待解决的问题[14, 15]。
微生物的样本通常是收集受试者的粪便,并被随机用于下游测序分析。研究人员发现,DNA提取前样本的同质化会影响样品之间的变异度[15, 16]。然而,尚无数据表明样本的不同采样点会在多大程度上影响WMS的测序结果。
随着测序方法的发展,越来越多高通量、短周期和低价格的产品可供研究人员选择。Illumina公司生产的HiSeq 2500、MiSeq、HiSeq X10和最新发布的NovaSeq系列等测序仪在WMS的测序研究中具有压倒性价格和产量的优势。这些测序仪之间是否存在显著差异,以及哪种是当前宏基因组测序实践的最佳选择,仍有待进一步研究讨论。
近来,针对微生物分类学图谱的几项大型研究的结果在一定程度上阐明了这个问题。例如,微生物组质量控制计划(Microbiome Quality Control Project, MBQC)利用16S rRNA测序方法比较了不同的粪便样本保存方法、DNA提取试剂盒、序列长度和生物信息学工具,并指导了肠道微生物组研究的实验设计[17]。另外,Costea等[15]比较了21种DNA提取试剂盒,并推荐了用于人类粪便样本的标准化方法。Walker等[18]的工作主要集中在16S rRNA PCR引物的选择,以及测序结果与荧光原位杂交法(fluorescent in situ hybridization, FISH)间的比较。这些研究主要关注基于16S扩增的测序方法的比较,但基于WMS的测序方法仍被忽视。
SRA数据库中已上传超过179万条“16S”和220万条“宏基因组”序列(截至2021年8月17日)。然而,生成WMS数据库的规模趋势正在以惊人的速度增长。由于16S rRNA基因测序和宏基因组鸟枪测序基于各自独特的方法,以及获得不同方面的信息,这可能会进一步导致对结果的错误解释[18, 19]。16S方法可识别和量化存在于所有细菌和古细菌中的标志基因rRNA,并使用现有的大型公共数据集进行比较;而WMS法则测量样本内所有生物体的整个基因组。过去,人们对16S和WMS方法进行了一些研究,例如文库试剂盒[20]之间的描述性比较,或使用样本的α和β多样性来评估群体内受试者之间的差异[8]。然而,目前利用配对样本直接比较16S和WMS之间的分类图谱研究尚为缺乏,仍然存在两者数据集能否在同一分析中进行比较的问题。
受微阵列质量控制(microarray quality control, MAQC)、测序质量控制(sequencing quality control, SEQC)和新发布的MBQC项目的启发,本文利用来自4个健康供体粪便样本的64个测序数据样本,研究样品同质化、文库制备方法和测序仪等因素的可比性问题[17, 21-23]。从一个大样本的不同采样点收集生物学重复,并使用WMS方法评估3种广泛使用的测序仪(HiSeq 2500, HiSeq X10和NovaSeq 6000)。本文还应用HiSeq 2500这一测序仪比较了16S扩增子和WMS文库制备方法。最后,本文构建了一种算法来提高16S和WMS方法之间的可比性。
1 材料与方法
1.1 样品收集和DNA提取
本研究从复旦大学泰州健康科学研究院的泰州队列中采集供体A、B、C、D 共4份粪便样本。简单来说,从4个样本中每个收集2个独立的取样点作为生物学重复。同时,每个生物学重复经过样品同质化产生2个相同的等分作为技术重复。16个样本收集后立即转移到-80 ℃冰箱中。本研究获得了所有4名捐赠者的知情同意。采用宏基因组DNA提取试剂盒(Tiangen, China)和溶菌酶(Sigma-Aldrich, Canada)对DNA标本进行纯化,最终产生了16个WMS文库和16个16S文库。
1.2 16S rRNA基因文库制备
本研究采用了V3-V4引物集,该引物集在16S测序研究中受到了广泛应用[23, 24]。根据Illumina的说明书设计并合成16S rRNA基因V3-V4扩增引物(插入片段469 bp, V3-V4扩增片段536 bp,编码文库613 bp)。正向引物337F: 5′-TCGTCGG CAGCGTCAGATGTGTATAAGAGACAGCCTACGGGN GGCWGCAG-3′;反向引物805R: 5′-GTCTCGTG GGCTCGGAGATGTGTATAAGAGACAGGACTACHVG GGTATCTAATCC-3′。引物稀释后,在文库制备前保存于-80 ℃条件。将12.5 ng微生物DNA、扩增子引物和HiFi HotStart ReadyMix (KAPA, USA)混合,再进行V3-V4 PCR试验。热循环条件包括95 ℃ 3 min;循环25次的95 ℃ 30 s、 55 ℃ 30 s和72 ℃ 30 s;72 ℃ 5 min和4 ℃保温。使用V3-V4扩增子反应的产物对用于Illumina测序的编码库PCR进行测序。利用N7和N5指数、95 ℃ 3 min、循环8次的95 ℃ 30 s、55 ℃ 30 s和72 ℃ 30 s;以及72 ℃放置5 min和保温4 ℃的PCR条件下生成独立文库。用1.1 × AMPure XP beads(Beckmann Coulter, USA)纯化文库。所有文库均通过Qubit 3.0和电泳定量或定性质量控制,并进一步以摩尔浓度1∶1、最终浓度2 nmol/L汇集。
1.3 全宏基因组鸟枪法文库制备
首先使用1 ng的微生物DNA片段进行分裂(Nextera XT试剂盒,Illumina,USA)。再使用NPM聚合酶(Illumina)和Illumina N5和N7指数特异性引物进行12个周期的PCR扩增DNA片段。文库纯化使用0.8 × AMPure XP beads(Beckmann Coulter, USA)。经2100QC和Qubit定量的文库图谱分析,文库以摩尔浓度为1∶1、最终浓度为2 nmol/L汇集。
1.4 二代测序技术
16S文库使用Illumina HiSeq 2500测序仪(Illumina, San Diego, USA)上的250 bp双端读流单元进行测序。WMS文库使用HiSeq 2500测序仪的250 bp双端读流单元,HiSeq X10和NovaSeq 6000测序仪的150 bp双端读流单元进行测序。供体B的1个样本在HiSeq X10编码中失败,未纳入分析。
1.5 生物信息学
所有64个原始FASTQ文件首先用FASTQC进行分析,以修剪过滤低质量的碱基。共有的读数使用PANDAseq和zcat命令拼接。16S文件用MALT分析生成RMA6格式文件,同时WMS文件用DIAMOND软件计算生成DAA文件。使用MEGAN 6包对指定读数的门、纲、目、科、属和种水平进行测定,生成不同种系水平每个样本的计数表。使用PANDAseq对16S配对端序列进行组装,确定共有数量,通过对共有区域的错误进行修正,重构并输出整个序列[25]。将组装好的长读数进一步使用MALT进行处理,将读数与SILVA rRNA数据库(Release v128a)进行比对以用于分类分析。利用zcat命令行对WMS的对端序列进行组合以获得长读数。当长读数生成后,使用DIAMOND工具将读数映射到NCBI参考宏基因组数据库[26]。相关、聚类和LEfSe分析中使用的分类图谱(WMS生成)是通过对非原核物种进行过滤处理得到。
1.6 经验贝叶斯方法
批次效应、测序仪和文库制备法的不同是微阵列研究领域中遇到的常见问题,特别是当组合来自不同实验的多批数据或实验不能同时进行时。查阅回顾既往文献,本文发现现有的调整方法,例如奇异值分解(single value decomposition,SVD)和距离加权判别法(distance weighted discriminant,DWD)不适用于处理批次大小很小的样本。由此,本文开发了一种经验贝叶斯方法,(empirical Bayes, EB)用于提高16S和WMS方法[27]之间的群体可比性。EB方法已被广泛应用于微阵列数据分析,特别当样本规模较小(<25)时,它能够稳健地处理高维数据[27]。EB法主要是在计算中“借用信息”,利用标准化数据和(非)参数先验分布估计批次效应参数,以期得到更好的估计或更稳定的推断。本文将EB法推广到调整微阵列数据的批次效应和提高文库制备的可比性问题中,对16S和WMS检测到的共有分类单元进行方法效应校正,以避免仅由一种方法检测到的分类单元所导致的校正算法的偏倚。用Yijt表示方法i中样本j的第 t种分类单元的表达式值,假设:
Yijt=αt+Xβt+γit+δitεijt
(1)
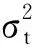
(2)
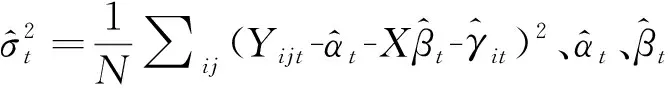
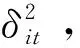
(3)
1.7 统计学分析
为了确定不同的提取方法之间哪些物种丰度有显著差异,对2个样本至少2个方案中丰度非零的物种应用了Bray-Curtis检验。考虑到多重检验问题,对结果的P值进行Bonferroni校正,校正后P值低于0.05被认为有统计学意义。Shannon Weaver指数、Simpson倒数指数、PCoA和Bray-Curtis矩阵使用MEGAN6(版本6.10.3)计算。使用R (v3.5.0)软件绘制Pearson相关性、Whiskers、相关热图和聚类图。采用LEfSe进行线性判别分析(LDA)。
2 结果
2.1 实验设计
粪便样本由来自泰州队列[28]的4个健康捐赠者(A、B、C和D)提供,提取标本中的DNA 用于文库制备。最终产生了用于WMS库和16S库的64个测序数据样本(Fig.1A)。16S样本进一步通过HiSeq 2500测序仪进行测序,而WMS样本采用HiSeq2500、HiSeq X10和NovaSeq 6000三个测序仪进行测序。16S样品采用PANDAseq[25]和MALT流程处理,而WMS则采用DIAMOND流程[26]。1个基于HiSeq X10的测序结果由于条形码技术故障而丢失,最终63个样品通过数据质量控制。
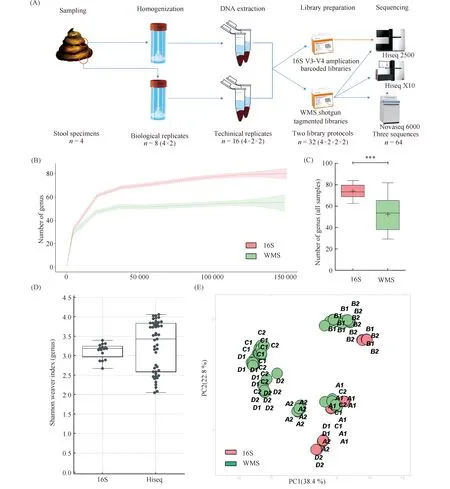
Fig.1 Experimental design and data description (A) Flow chart of library preparation from 64 samples. (B) The rarefaction curves of sequenced 16S (n=16) and WMS (n=48) samples. (C) Number of average detected genera by 16S (74.16±1.69) was significantly larger than that of WMS protocol (52.46±2.19). *** P < 0.001. (D) Shannon weaver index of genus level between 16S and WMS protocol had no statistically significant differences (P>0.05). (E) Bray-curtis distance differences between 16s and WMS based on PCoA
2.2 16S和WMS数据概述
计算了每个样本的稀疏度,并绘制了校准读数和检测到的属级数量之间的关系。结果表明,样品的测序结果到达了平台期,这保证了进一步分析数据的可靠性(Fig.1B)。16S方法检测到的平均属级数量(n=74.16±1.69)显著多于WMS方法(n=52.46±2.19)(Fig.1C,P<0.001)。在Shannon Weaver指数(α多样性)方面,来自16S和WMS方法样本间没有发现统计学差异(Fig.1D)。然而,基于Bray-Curtis距离(β多样性)的PCoA图显示了方法不同引起偏倚,这导致16S方法的聚类占优势,特别是在供体A、C和D中(Fig.1E)。然而,当从UPGMA、PCoA和N-J树中考虑WMS或16S结果时,大多数样本显示出供体来源的聚类。在WMS方法中,HiSeq 2000、NovaSeq 6000和HiSeq X10三个测序仪的结果紧密地聚类于技术重复,也就是说有些样品是根据它们的技术重复而不是生物学重复聚类的。
2.3 使用WMS进行测序仪的可重复性和样本的异质性分析
本文采用Shannon Weaver指数对3个使用WMS的测序仪在门、纲、目、科、属5个水平上进行了比较。测序仪在各等级之间未发现显著性差异。等级越低,α多样性越高(Fig.2A)。
为了确定粪便样本的异质性,使用Shannon指数对配对技术重复和生物学重复进行了比较。与生物学重复(Fig.2C, Pearsonr=0.69)相比,WMS方法显示技术重复具有更高的相关性(Fig.2B, Pearsonr=0.94)。
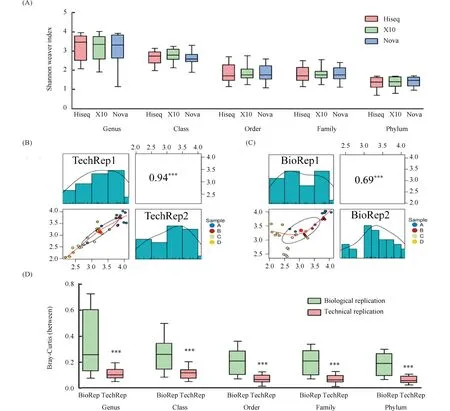
Fig.2 Sequencer reproducibility and Intra-specimen heterogeneity analysis using WMS (A) Shannon Weaver index of three WMS sequencers in five hierarchical ranks were calculated and compared. (B,C) Technical replicates and biological replicates in WMS protocol were compared using Shannon index and Pearson correlation coefficient. ***P<0.001. (D) Bray-Curtis index of the technical replicates and biological replicates in all five hierarchical ranks. ***P<0.001
采用Bray-Curtis 指数分析不同取样点(生物学重复)与同一取样点(技术重复)之间的差异。配对Bray-Curtis距离的结果显示,在所有5个等级中,3个测序仪的生物学重复差异大于技术重复(Fig.2D,P<0.001)。仅使用HiSeq 2500测序仪的16S rRNA基因测序数据中也显示类似的结果(P<0.05)。本文进一步使用基于LDA的方法计算每个测序仪的丰度差异物种。结果显示,无特殊的物种或测序仪,表明不同测序平台测序结果具有一致性和可重复性。
2.4 文库制备方法不同产生的差异
本文使用α多样性进一步比较测序方法、测序仪在样本间的差异。由WMS方法间的Pearson相关性,结果显示,测序仪之间具有高度的一致性(Fig.3A)。然而,使用HiSeq测序仪的16S和使用3种测序仪的WMS方法之间的Shannon指数存在巨大的差异,尤其是在样本A、B和D之间(Fig.3A,B)。
基于Bray-Curtis距离,本文发现,在属水平上16S与WMS方法之间的差异远远大于技术重复、生物学重复或测序仪间的距离(Fig.3C,P<0.001)。令人惊讶的是,测序方法间的Bray-Curtis差异指数与独立样本间一样大(0.59±0.05vs0.64±0.01,P=0.22)。
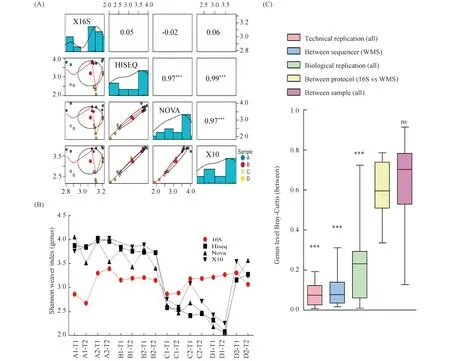
Fig.3 Library preparation induced dissimilarity (A) Comparison of paired samples using alpha diversity across 2 protocols and 3 sequencers. There was a higher consistence among sequencers than in protocols. (B) Shannon Weaver index of dissimilarities between 16S and WMS. The difference was large especially in sample A, B and D. (C) Compared with the dissimilarity between technical replicates, sequencers, biological replicates, protocols or samples. ns, not significant. ***P < 0.001
2.5 16S和WMS方法中的特定分类学图谱
本文首先比较了所有5个等级中16S和WMS间的配对分类图谱。与高级别分类相比,属水平的Pearson相关性较低(Fig.4A)。为了进一步揭示在属水平中检测到的偏倚特征,本文绘制了维恩图,以显示每个测序仪和方法检测到的共有部分以及独特的分类单元(Fig.4B)。
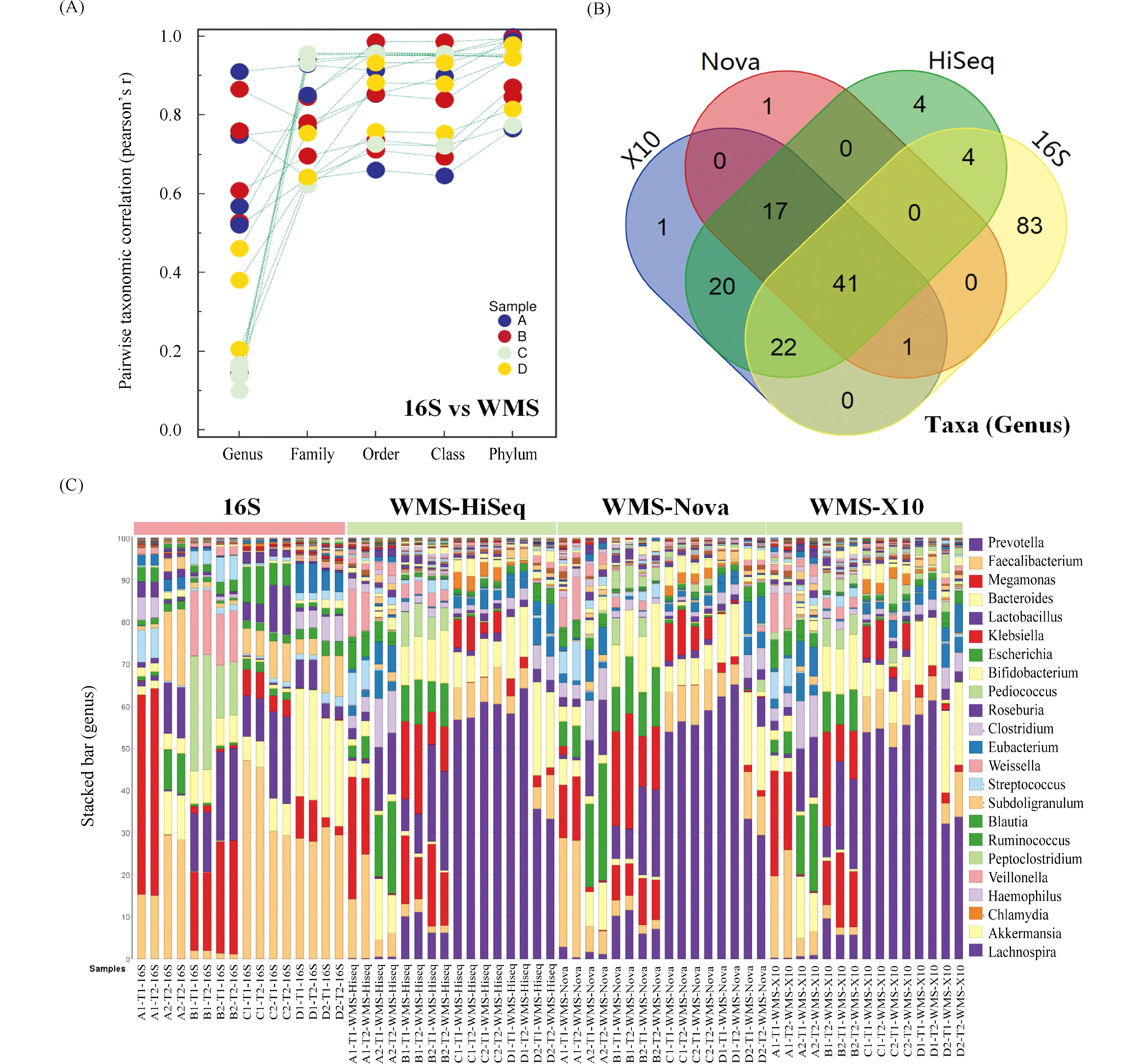
Fig.4 Specific taxonomic profiles in 16S and WMS protocols (A) Pairwise taxonomic assignments between 16S and WMS protocol were compared with Pearson’s correlation. (B) Venn diagram presented overlapping and unique taxa by each sequencer and protocol. (C) Stacked bar plot exhibited all samples’ taxonomic assignments in the genus level
样品的条形图显示,16S方法中Faecalibacterium(粪杆菌)和Megamonas(巨单胞菌)占优势菌,而WMS样品则倾向于以Prevotella(普雷沃氏菌)为优势菌(Fig.4C)。16S方法显示,83个属水平的独特类群(计数>1),包括Kosakonia(科萨克氏菌)、CandidatusSoleaferrea(瘤胃菌科)和Peptococcus(消化球菌属), 而WMS鉴定了70个特定属,包括Dialister(小杆菌属)、Olsenella(欧陆森氏菌属)和Akkermensia(阿克曼氏菌属)。除了独特的检出类群外,有偏特征显示出它们对2种制备方法之一的偏好,这也可能导致了不可重复性。本文进一步利用HiSeq 2500数据,应用线性判别分析检测每个方法的人工生物标志物[29]。WMS对Clostridia(梭菌)和Chlamydia(衣原体)的鉴定效果较好,而16S方法对Cyanobacteria(蓝藻菌)和Rhodobacteria(红藻菌)的鉴定效果较好。
2.6 提高群体可比性的算法
为了降低16S和WMS间的方法效应,本文提出了一种经验贝叶斯算法来提高群体可比性。结果表明,经验贝叶斯算法显著增强了16S和WMS数据集中共有属之间的方法学相关性,从r=0.45提高到r=0.70(Fig.5A)。在应用贝叶斯过程后,文库制备方法的差异特征被消除,微生物群特征趋于相似(Fig.5B)。此外,该算法降低了由16S方法导致的PCoA图的聚类偏倚,最终样本依据供体来源聚集 (Fig.5C)。最后,本文采用同样的方法对泰州队列另外2名健康捐赠者的粪便样本进行了测序,以验证所提出的贝叶斯方法的有效性。由通过质量控制的30个测序数据的结果,经过贝叶斯算法校正,微生物群特征趋于相似(从r=0.37提高到r=0.59),且PCoA聚类偏倚有所改善。据此,此贝叶斯算法能有效提高不同测序来源群体的可比性。
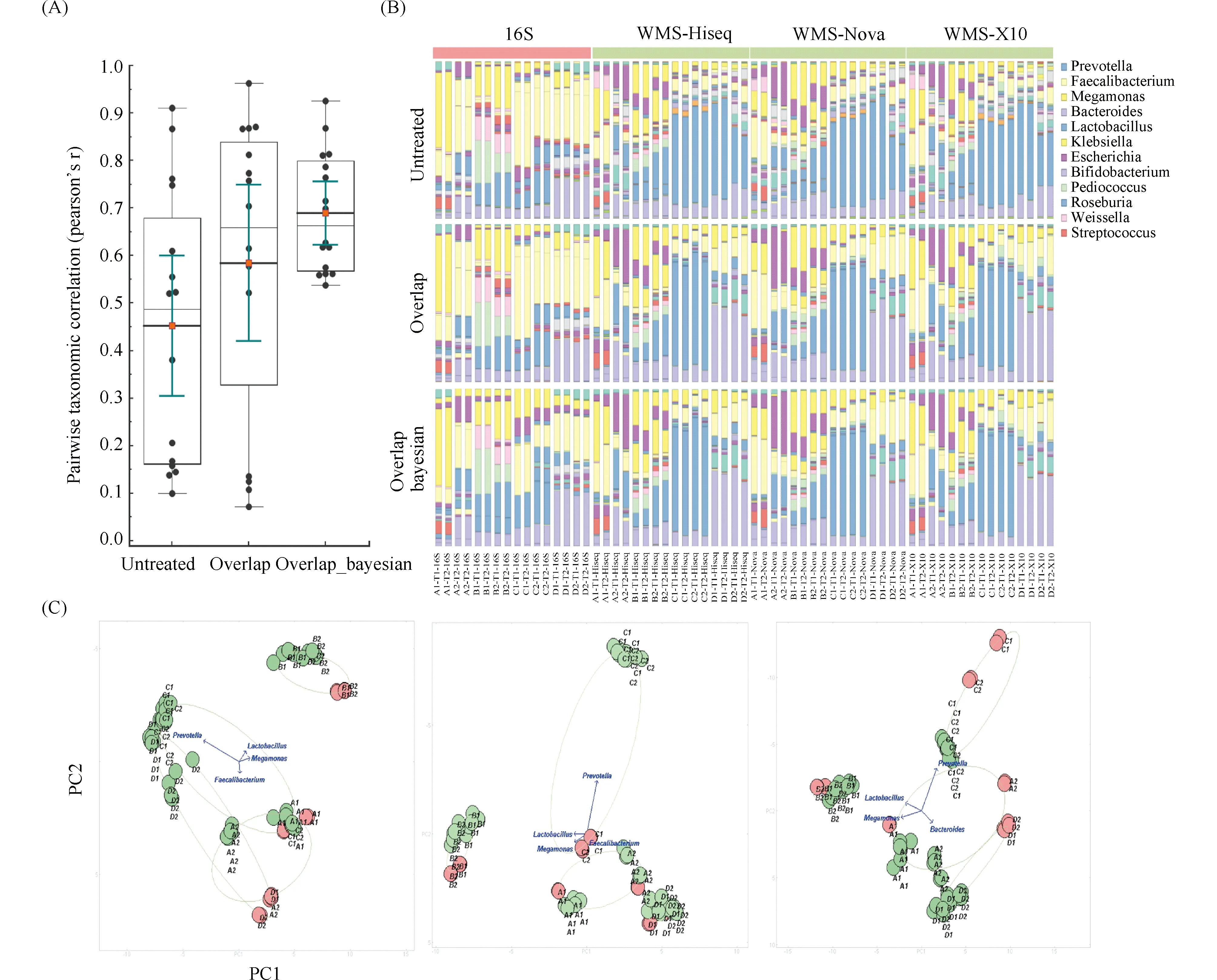
Fig.5 Empirical Bayesian algorithm to improve population-wide comparability (A) Pearson correlation coefficients of samples with 16S and WMS before and after using empirical Bayes algorithm. The algorithm enhanced the correlation in overlap genus level from r=0.45 to r=0.70. (B) Microbiota relative abundance and patterns in untreated, overlap and Bayesian stage. Differences in library preparation methods were eliminated. (C) The PCoA clustering plot based on 16S and WMS protocol from different donors. Almost all the samples are clustered by their donors after the Bayesian process
2.7 基于经验贝叶斯算法的外部数据集验证
本文应用经验贝叶斯算法融合分析了Dubin等[30]收集的10个WMS和16S配对肠道微生物样本(PRJNA302832)的数据集。本文的算法显著增强了样本间的相关性(Fig.6A)。基于Bray-Curtis的PCoA也显示,在消除了测序方法的偏倚后,同一供体的样本成功聚类(Fig.6B,C)。
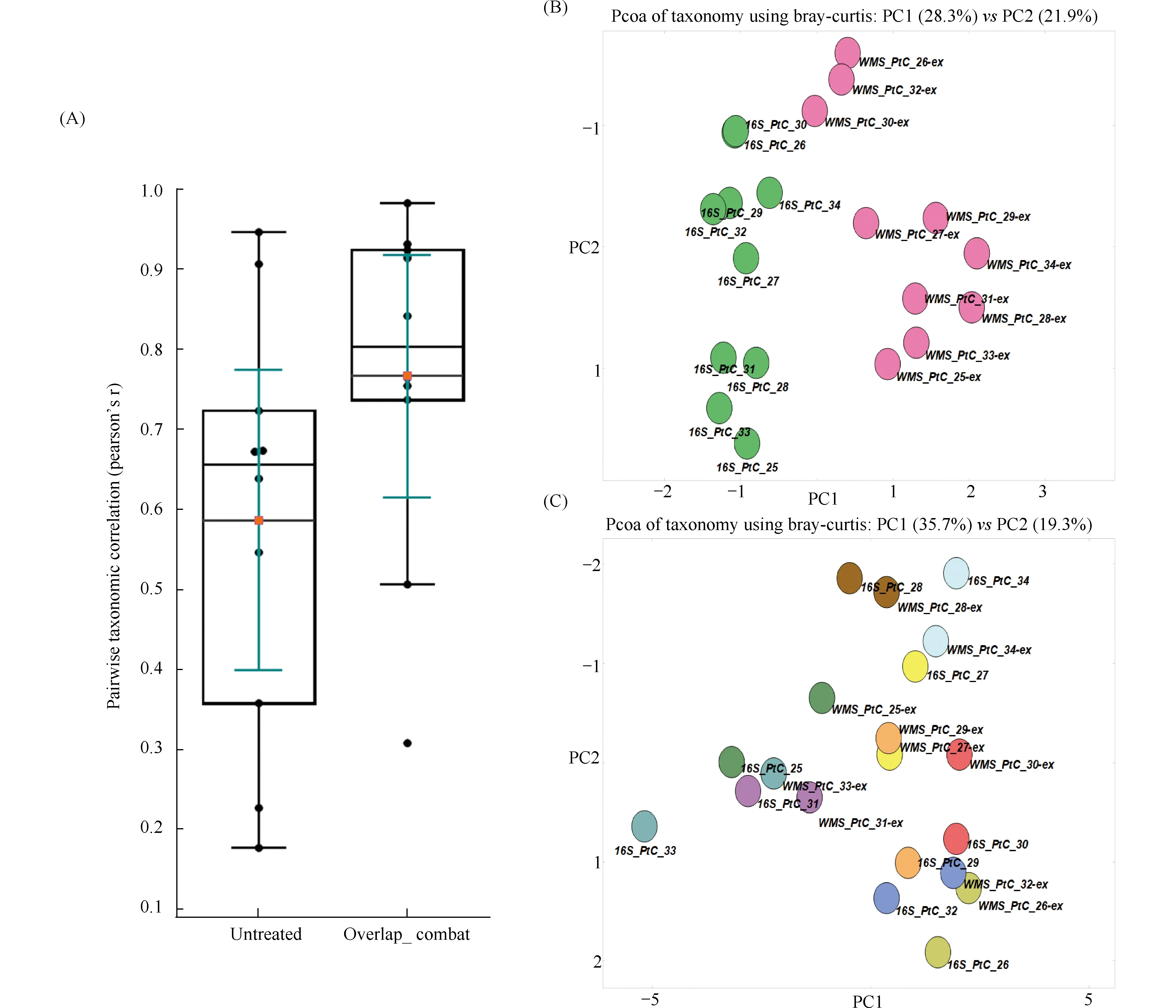
Fig.6 Validation of the Empirical Bayesian based algorithm We applied our algorithm to a published gut microbiota dataset with 10 WMS and 16S pairwise samples (PRJNA302832). (A) A significant enhancement of sample-to-sample correlations was obtained compared with untreated samples. (B,C) The samples were successfully clustered as their donor origin in the PCoA after protocol induced effect was removed
2.8 测序仪和文库制备的一些事实
最后,本文总结了肠道微生物组研究生成测序文库的最小成本。NovaSeq 6000向用户展现了最具成本-效益的性能和时间价值,而HiSeq 2500和HiSeq X10则需要花费更多的时间和成本。考虑到良好的重现性和Simpson多样性的要求,使用WMS文库制备法和NovaSeq测序仪是最优的选择。本文在一台配备128 Intel E7 4870 Quadcores, 1TB DDR4-2400 MHz, 10K rpm SAS 12Gb的服务器上测试并估计了其性能。
3 讨论
本文对样本的异质性、测序仪和文库制备方法等问题进行了肠道微生物测序的质量控制研究。结果证实,每个样本中的微生物组成在不同的取样点上是不同的。3种测序仪在多样性和分类丰度方面均得到了相似的测序结果。在16S和WMS方法生成的数据集中均能观察到独特的分类图谱。最终,本文设计并提供了一个有效的模型来增强两种测序数据之间的可比性。
同质粪便样品已被证明可为下游处理提供相同的试样,从而产生可重复的数据[15,16]。然而,以往的研究使用同质样本来分析多组学数据或横向比较多个变量,而不是显示样本本身异质性的影响。Codling等发现,粪便与粘膜样本之间存在差异,而Friswell等在胃肠道中发现了特异性位点的微生物群[31,32]。Hsieh等利用16S V4测序评估了微生物样本的取样点和处理方法[17,33]。与本文的结果类似,样本同质化显示出减少数据集中个体内变异的巨大潜力。同质化或多次取样对于减少下游测序的变异是必要的。
本文使用WMS方法比较了3种广泛采用的测序仪HiSeq 2500、HiSeq X10和NovaSeq 6000。它们的测序结果在生物学多样性和分类图谱方面基本一致,具有较好的可重复性。MBQC使用16S rRNA方法比较了HiSeq和MiSeq,表明测序仪引起的差异小于样本间差异,获得了与本文相似的结论[17]。NovaSeq 6000在运行数据量、每样本价格和测序速度方面,都比HiSeq 2500和HiSeq X10表现出压倒性的竞争力和更高的性价比。从NovaSeq 6000输出的数据高达1.5TB,这意味着通过多路传输可以同时测序500~800个样本。因此,当测序批次对测序结果有影响时,这将有利于控制批次效应。
MBQC等研究使用了大规模的盲法样本集,解决了人类微生物组测序方法、样本处理方法和微生物数据处理计算流程等问题[15,17,18]。正如Costea等所提出的,在过去5年中,超过3 000项研究(从环境到生物医学研究)对微生物群落进行了调查,产生了超过160 000份发表的16S和WMS宏基因组数据样本[15,34]。这些结果强调了生物信息学工具和DNA提取试剂盒的特异性,而本文的研究重点是测序平台、采样点之间的差异和文库制备方法的特异性。
本文的结果进一步表明,在所有量化的因素中,文库制备方法的差异对观察到的微生物组成的影响最大;这种影响甚至与样本间差异导致的同样大。16S rRNA测序是有偏差的,因为16S rRNA基因扩增不均匀,而WMS法可能因测序不够深导致无法检测到稀有物种[35]。Steven和Poretsky等的研究通过低维聚类或描述性比较揭示了16S基因测序与鸟枪测序的差异。然而,这些研究并未显示配对方法间的距离和相关性比较[36,37]。在更大规模的宏基因组质量控制研究中,人们已经揭示了16S和WMS方法之间的差异。在Sinha等人的研究中,16S与WMS之间的Spearman相关系数为0.57~0.74,表现出中度正相关[17]。本文的研究结果表明,两种测序方法的Pearson相关系数在0.2~0.8之间,主要受样本来源的影响。Jovel等[19]就16S和WMS方法间的多样性讨论了类似问题,结果表明,模拟细菌在使用16S和WMS时有检测偏倚。有趣的是,在组成更简单的细菌群落中,16S与WMS结果的相对丰度模式相似。当比较Simpson倒数指数在16S与WMS之间的相关性时,本文的数据支持这一结论。Hillmann等[38]在研究中,与16S方法相比,WMS的数据可以观察到许多特定的物种。与本文的结论类似,16S和WMS在属水平上的一致性高于种水平。
WMS在分类图谱中显示出更高的预测能力,即可以获得物种甚至菌株水平的信息,而16S在属水平上只能达到约80%的准确率[39,40]。16S和WMS可以相互补充,发挥各自的优势。16S方法适用于组织等含有或被高比例宿主DNA污染的样本[41],而WMS法不需要给定微生物群落,在粪便或唾液样本中均有效。因此,为了更好地提高16S和WMS方法之间的可比性,需要用算法来减少测序方法不同引起的偏倚。此前,在这一领域的生物信息学研究主要是尝试提高16S rRNA基因测序分类图谱的预测能力和系统进化率,例如COMPASS和EMIRGE[42-44]。本文设计的经验贝叶斯法在减少方法带来的差异方面以及在内外部数据集中都有较好的表现,特别是在处理使用相同测序仪的配对样本中,原因是这种方法将数据的不能消减效应视为加法和乘法效应,实际是一种均值中心化算法和尺度算法的混合[27]。
本文的研究在真阳性率和qPCR验证方面仍存在一些局限。然而,FACS或qPCR试验的分类丰度推断,也可能是由于杂交错误或多余PCR产物的过度扩增而导致的系统误差[18]。单分子和长读测序(例如MinION或PacBio)改进的宏基因组测序,有希望成为得到更精确分类图谱的替代方案[45]。
本文的研究结果表明,同质化是样品DNA提取前的必要步骤。测序仪对分类学变异的贡献小于文库制备方法。经验贝叶斯方法提高了16S和WMS之间的群体可比性,这意味着它在进一步融合分析已发表的16S和微生物数据集方面具有强大的潜力。
