内皮联蛋白在血管生成中的调控机制及作用
2022-09-07李炳轩
李炳轩, 黄 涛
(天津大学生命科学学院结构生物学实验室, 天津 300072)
血管生成(angiogenesis)是指在原有血管上生成新血管的生理过程,在机体发育、伤口愈合及促肿瘤恶化等方面有着重要影响[1]。由组织缺氧等引发的血管出芽(sprouting)是血管生成的基本方式:机体组织释放的血管内皮生长因子(vascular endothelial growth factor,VEGF)和转化生长因子β(transforming growth factor β,TGF-β)等会激活邻近血管内皮细胞的VEGF和TGF-β等信号通路,被激活的内皮细胞会向外增殖延伸形成“芽”,并发育成新血管[2]。
在参与血管生成的细胞因子中,TGF-β家族蛋白质极为重要。该家族中有3种受体,包括激活素受体样激酶1(activin receptor-like kinase 1,ALK1)及激活素受体样激酶5(activin receptor-like kinase 5,ALK5)等在内的I型受体,转化生长因子受体II(TGFβ receptor II,TβRII)及骨形态发生蛋白受体II(bone morphogenetic protein receptor II,BMPRII)等在内的II型受体,以及内皮联蛋白(endoglin,ENG)等在内的辅助受体[3]。它们通过一套固定的信号传导模式调控着内皮细胞的命运:在辅助受体的参与下,TGF-β家族蛋白质与细胞膜上的I和II型受体互作而形成三元复合物,致使受体胞内域被磷酸化,然后信号经胞内SMADs依赖型或SMADs非依赖型两条信号通路传递,并最终调控基因转录,转录出的蛋白将进一步调控血管生成[4]。其中,SMADs依赖型通路主要由SMAD1~9等因子组成,且依赖于I型受体的磷酸化;SMADs非依赖型通路由多种因子组成,是多条信号通路的统称[3, 5]。
ENG是TGF-β家族蛋白质的辅助受体,是一个主要表达于内皮细胞的分子质量约为90 kD的I型跨膜糖蛋白。此外,ENG亦在成纤维细胞与血管平滑肌细胞等细胞中表达[6-9]。ENG通过直接或间接影响上述细胞的增殖、迁移、黏附或分化等生理作用调控血管生成,其异常可能会导致血管畸形和癌症恶化等后果:已鉴定出100多种Eng基因突变会引发一种以血管发育不良、动静脉畸形和反复出血等为特征的1型遗传性出血性毛细血管扩张症(hereditary hemorrhagic telangiectasia 1,HHT1)[10];实体瘤血管内皮细胞会高表达ENG,以促进肿瘤血管生成与癌细胞转移[11];Eng纯合子缺失型小鼠会因血管网络发育失败而死于胚胎期[6]。ENG之所以能够在血管生成中发挥重要功能,是源于ENG独特的结构,而其结构特征又决定了ENG能够与不同蛋白质互作来调控血管生成。本文分别从ENG与TGF-β及非TGF-β家族相关蛋白质的互作入手,总结了ENG调控血管生成的机制,以期对今后ENG相关研究有所帮助。
1 内皮联蛋白的结构
人源内皮联蛋白包含1个较大的胞外域(561个残基)、1个跨膜域和1个较小的胞内域(47个或14个残基)[7](见Fig.1)。
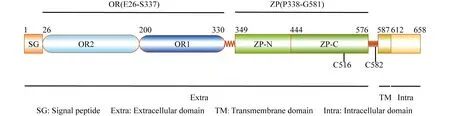
Fig.1 Schematic diagram of ENG structure ENG contains an extracellular domain, a transmembrane domain and a relatively small intracellular domain. The extracellular domain contains signal peptide, OR and ZP. OR contains OR1 and OR2. ZP contains ZP-N and ZP-C. C516 and C582 are two free cysteines
1.1 ENG的胞外域
ENG的胞外域包含orphan domain(OR)和透明带域(zona pellucida domain,ZP),OR由OR1与OR2组成,ZP由ZP-N与ZP-C组成,这4部分均以堆叠的β片层为基本骨架,形成免疫球蛋白样的结构[7]。OR的晶体结构(PDB ID:5I04)显示,OR1上的β6~β7片层组成了一个疏水沟,负责结合配体骨形态发生蛋白9(bone morphogenetic protein 9,BMP9)[12]。虽然OR1与OR2在结构上高度相似,但是与OR1结合BMP9的功能不同,目前尚未发现OR2具有任何介导蛋白质互作的功能,因此关于OR2的功能仍需要进一步研究[12]。ZP-N包含1个Arg-Gly-Asp(RGD)序列,这是一个结合整合素的经典序列,ZP-C包含的C516及近膜端包含的C582,分别形成C516~C516和C582~C582共2个二硫键并共同介导ENG的二聚化[12, 13]。
1.2 ENG的胞内域
细胞通过选择性剪接ENG的前体mRNA (precursor mRNA)产生两种亚型。依据胞内域的长度,ENG可被分为长型-ENG(L-ENG,胞内域含47个氨基酸残基)与短型-ENG(S-ENG,胞内域含14个氨基酸残基)[14, 15]。当内皮细胞处于健康活跃状态时,L-ENG为主要亚型,而当内皮细胞处于衰老或高氧状态时,S-ENG会被大量诱导表达,从而降低内皮细胞的增殖率,抑制血管生成[14, 16]。L-ENG胞内域含有丝/苏氨酸富集区,该区域可被I型或II型受体磷酸化,其C端Ser-Met-Ala 的3个氨基酸是一个I型PDZ结合序列,可结合其他蛋白质的PDZ结构域。相反,S-ENG的胞内域因缺少这些特殊序列,所以无类似L-ENG的功能[17]。ENG的两种亚型具有截然不同的生理功能,L-ENG可帮助内皮细胞迁移或增殖,S-ENG可参与内皮细胞衰老[14]。
2 内皮联蛋白与TGF-β家族相关蛋白质互作调控血管生成
2.1 ENG与TGF-β家族的I型受体互作调控血管生成
L-ENG和S-ENG分别与ALK1及ALK5互作来介导胞内2条功能不同的SMADs信号通路,进而改变TGF-β信号传导的结果[14](见Fig.2)。L-ENG胞内域与ALK1的亲和力较高,可触发由被磷酸化的ALK1引起的SMAD1/5信号通路。已有研究表明,ALK1-SMAD1/5的激活可以增加VEGF的表达量,并促进血管生成等[14, 18];S-ENG胞内域与ALK5的亲和力较高,可促使TGFβ1等因子的信号经由ALK5-SMAD2/3通路传递,而ALK5-SMAD2/3信号通路可以调控内皮细胞与血管平滑肌细胞,来分泌结缔组织生长因子(connective tissue growth factor,CTGF)等促进细胞外基质形成的因子,进而调控细胞外基质稳态与血管形成的过程,而这些因子的过表达则可能导致血管纤维化等血管病变[14, 19-21]。在细胞衰老阶段,血管中的CTGF表达量被上调,这导致血管细胞外基质发生重塑,并进一步诱发衰老相关的血管病症等[19]。
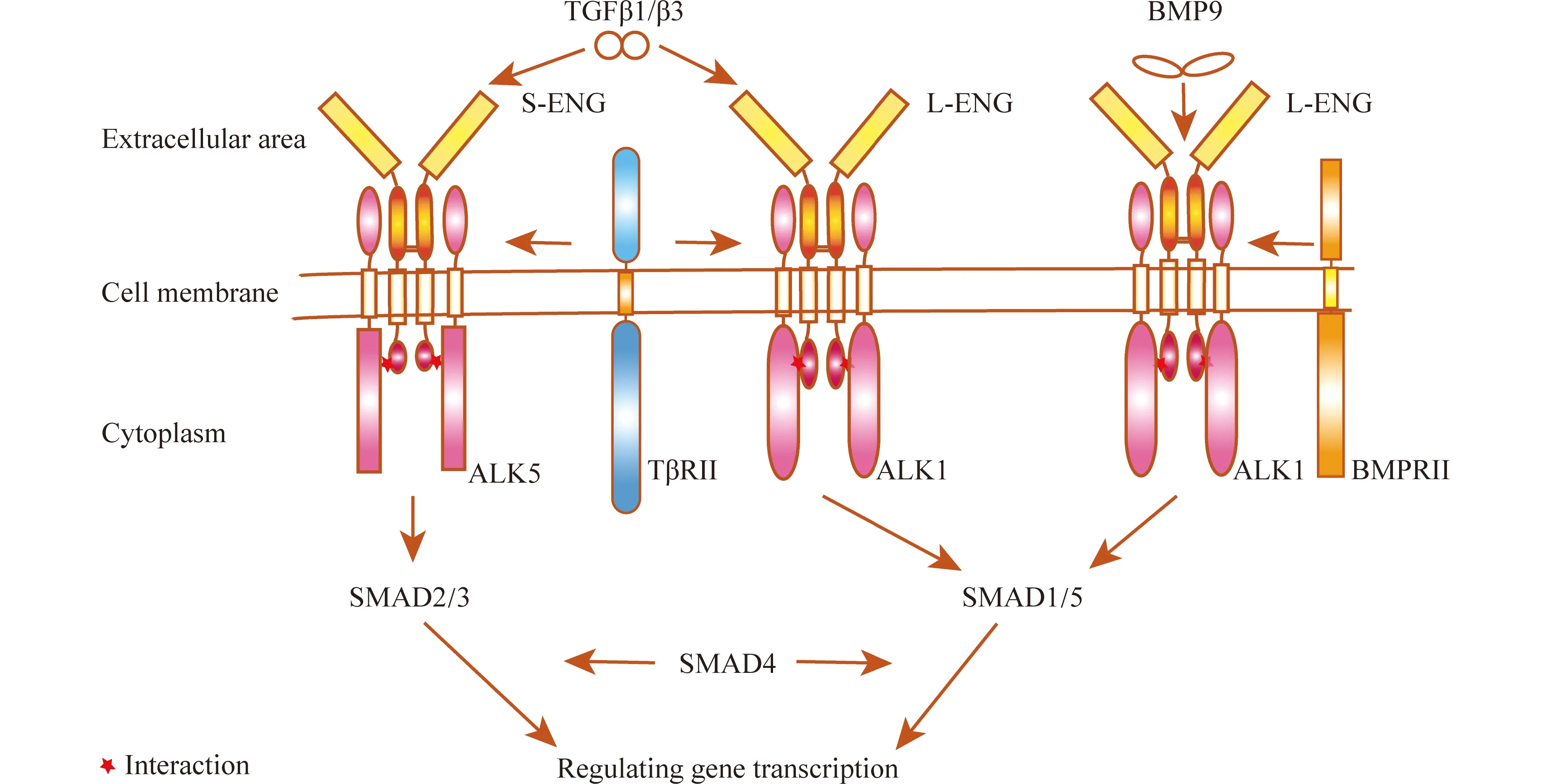
Fig.2 Diagram of ENG signaling pathways TGF-β1/β3 bind to ALK5 or ALK1 with the assistance of S-ENG or L-ENG, subsequently bind to TβRII. BMP9 binds to ALK1 with the assistance of L-ENG, subsequently binds to BMPRII. ALK5 will activate SMAD2/3 and ALK1 will activate SMAD1/5. With the assistance of SMAD4, these SMADs factors will enter the nucleus to regulate the transcription of corresponding genes related to angiogenesis
此外,ALK1和ALK5亦可影响ENG的功能。Tazat等[22]发现,被磷酸化的ALK1与ALK5会被胞吞,但ALK1被胞吞的速率(t1/2=2.5 min)远高于ALK5(t1/2=13 min)。同时,ALK1可通过与ENG形成复合物使ENG一同被胞吞,进而抑制SMAD1/5信号通路,抑制血管生成。
2.2 ENG与TGF-β家族蛋白质互作调控血管生成
ENG可与TGF-β家族蛋白质TGFβ1/β3相互作用,调控涉及TGFβ1/β3的血管生成过程[23](见Fig.2)。TGFβ1是一个经典的促进细胞纤维化的因子,Kapur等[24]发现,通过降低心力衰竭患者的左心室内皮细胞中ENG的表达量能抑制TGFβ1的信号,限制心肌纤维化并提高心力衰竭患者的存活率,说明ENG在TGFβ1信号通路调控心血管发展过程中发挥着重要作用。
后续研究发现,ENG与TGF-β家族蛋白质BMP9的亲和力远高于ENG与TGFβ1的亲和力[12],ENG可通过与BMP9互作来调控血管生成(见Fig.2)。基于二者的蛋白质互作结构已被解析,ENG与BMP9的互作界面包括ENG-OR1中的1个高度保守的疏水沟和BMP9生长因子中的β6~β7片层[12]。ENG在结合BMP9时亦可以竞争性替换掉BMP9的前体结构域,使BMP9暴露出结合II型受体的位置[12, 25]。研究显示,I型与II型受体均不能替换掉BMP9前体结构域,且II型受体与BMP9结合界面被BMP9的前体结构域阻碍,因此ENG对BMP9-I型受体-II型受体的三元复合物的形成至关重要[26, 27]。
3 内皮联蛋白与非TGF-β家族相关蛋白质互作调控血管生成
3.1 ENG与MMP14互作调控血管生成
基质金属蛋白酶14(matrix metalloproteinase 14,MMP14)可识别并切割ENG的G586与L587间的肽键,产生一种不包含跨膜域与胞内域的游离型ENG。因此,血液中游离型ENG的含量与MMP14的含量呈正相关,在临床上游离型ENG含量的升高通常伴随着子痫前期或肺动脉高压等病症[28-30]。目前,游离型ENG调控血管生成进而导致这些疾病的机制仍存在争议:Castonguay等[31]发现,外源游离型ENG-Fc重组蛋白能特异性结合配体BMP9/10,并抑制A204细胞系(该细胞不含内源ENG)对2个配体的响应。但Lawera等[25]发现,当人肺动脉内皮细胞表达内源ENG时,外源游离型ENG单体不会抑制细胞对BMP9的响应;当人肺动脉内皮细胞不表达内源ENG时,外源的游离型ENG单体可抑制BMP9/10的信号。因此,MMP14可以通过产生游离型ENG,调控BMP9/10信号通路进而调控血管生成,但具体机制可能涉及多种因素,目前尚不明确。
3.2 ENG与整合素互作调控血管生成
整合素是一种由α亚基和β亚基组成的异源二聚的膜受体[32]。整合素可通过与包含RGD序列的配体互作,进而激活黏着斑激酶(focal adhesion kinase,FAK)和磷脂酰肌醇3-激酶(phosphatidylinositol 3-kinase,PI3K)等,最终调控血管生成[32, 33]。α5β1是一种在内皮细胞中表达的整合素,其主要配体为纤维粘连蛋白(fibronectin,FN),但亦能与包含RGD序列的ENG等互作,强化ENG促进血管生成的功能[34, 35]。
细胞外基质中的纤维粘连蛋白可通过与α5β1互作使α5β1在细胞表面聚集成簇,而成簇的α5β1可以与ENG、ALK1互作,进而促进ENG与ALK1形成复合物,增强促血管生成的SMAD1/5信号;同时,TGFβ1也能够以依赖ENG,但不依赖I型受体的方式激活α5β1信号通路,TGFβ1对α5β1信号的激活依赖由β-抑制蛋白2(β-arrestin2,βarr2)引起的ENG被胞吞的生理活动,这使与ENG胞外域互作α5β1亦被胞吞,进而激活FAK通路,抑制内皮细胞迁移,最终参与促进新生血管稳定与成熟[35, 36]。
3.3 ENG与LRG1互作调控血管生成
正如上文所述,有多种蛋白质可以通过与ENG互作进而影响TGF-β通路来调控血管生成,但ENG参与到病理性血管生成的机制却尚不明晰。近年来,研究发现一种名为富含亮氨酸的α-2-糖蛋白-1(leucine-rich alpha-2-glycoprotein-1,LRG1)的血管生成性糖蛋白,相比于野生型小鼠,高表达LRG1的小鼠眼部的病理性血管的数量增加,而Lrg1基因敲除小鼠眼部的病理性血管的数量减少,同时正常血管的数量无明显变化。这表明,LRG1可能在病理性血管的生成中发挥主导作用,而在正常的血管生成中作用不明显[37]。后续的生化研究表明,在病理状态下,高表达的LRG1可与ALK1或ALK5结合。但在TGFβ1及ENG存在时,LRG1则通过结合ENG,增强与ALK1的互作,削弱与ALK5的互作,这导致ALK1-Smad1/5信号被增强,从而促进病理性血管的生成[37]。LRG1是一个具有重要意义的发现,使在不损伤正常血管生成基础上抑制病理性血管生成变为可能,但由于目前ENG与LRG1的蛋白质互作界面尚未解析,具体作用机制尚不明了。因此,未来研究可以尝试通过解析ENG与LRG1的互作结构来开发相应的靶向药物。
3.4 ENG与VEGFR2互作调控血管生成
VEGF及其主要受体(VEGF receptor,VEGFR)是促血管生成的关键因素。VEGF有A、B、C、D和胎盘生长因子5个亚型。其中,VEGF-A及其受体VEGFR2在血管生成阶段发挥关键作用[38, 39]。VEGFR2在结合VEGF后会被胞吞,但ENG的胞内域序列625PVVAVA630和643SIGSTQ648与VEGFR2以VEGF-A依赖性方式互作,因此ENG可以抑制VEGFR2被胞吞,延长VEGFR2在细胞膜上的驻留时间(驻膜时间),促进血管出芽与延伸[38, 39]。然而,缺失胞内域的ENG则不能结合VEGFR2,这导致VEGF-A信号半衰期被缩短,出芽活动被削弱,血管向外延伸能力下降,同时血管局部增殖并导致血管畸形[39, 40]。目前在小鼠模型中,联用VEGFR的单克隆抗体bevacizumab与ENG的单克隆抗体TRC105,可以成功抑制注射进小鼠体内的4T1癌细胞的生长与转移,这为通过联合用药抑制肿瘤的血管生成提供了一定的理论与实践基础[39]。
3.5 ENG与支架蛋白互作调控血管生成
G蛋白信号调节体-G alpha相互作用蛋白C端(GAIP interacting protein C-terminus,GIPC)是一个包含PDZ结构域,可以稳定跨膜蛋白质的支架蛋白[41]。ENG可通过与GIPC互作来增加自身的驻膜时间。Lee等[17]发现,GIPC可通过与ENG胞内域的I型PDZ结合基序互作,稳定小鼠胚胎内皮细胞表面的ENG,延长ENG调控其他信号通路的时间,促进SMAD1/5的磷酸化,抑制血管内皮细胞的迁移,进而调控血管生成。
ENG亦可通过与另一种支架蛋白质,即G蛋白偶联受体信号通路的负反馈因子β-arrestin2互作引发ENG被胞吞,缩短ENG驻膜时间,并激活SMADs非依赖型通路,削弱细胞外调节蛋白激酶(extracellular regulated protein kinases,ERK)信号,最终抑制内皮细胞的有丝分裂和迁移。利用siRNA沉默内源β-arrestin2可恢复细胞的ERK信号与内皮细胞的迁移功能,进而调控血管生成。后续的研究进一步发现,ENG与β-arrestin2的互作依赖于ENG的T650附近区域的磷酸化。鉴于T650附近区域的磷酸化又依赖于TβRII或ALK1的磷酸化,因此ENG与β-arrestin2的互作可能涉及了ENG信号的负反馈调节机制[36, 42]。这项发现可能为治疗因ENG过表达而导致的肿瘤等疾病提供了新思路。但由于目前ENG与β-arrestin2复合物结构尚未解析,相关靶向药物的开发仍需更多的研究。
3.6 ENG与血管内皮钙黏连蛋白互作调控血管生成
血管内皮钙黏连蛋白(vascular endothelial cadherin,VE-cadherin)是一种表达于血管内皮、负责调控细胞黏连的Ca2+依赖性的膜蛋白质,参与血管生成与稳态调控等[43]。正常表达的且在细胞膜上规则分布的血管内皮钙黏连蛋白可强化细胞间黏连,减弱血管的渗透性[44]。ENG可通过与血管内皮钙黏连蛋白互作来改变其在细胞膜上的分布情况,并借以调控内皮细胞间的黏连。野生型小鼠和过表达ENG的小鼠的血管内皮钙黏连蛋白表达量虽无明显区别,但过表达ENG小鼠的血管内皮钙黏连蛋白倾向于不规则地分布于细胞膜上,这导致内皮细胞间的黏连被减弱,血管的渗透性增加,最终致使新生血管不稳定,易出现出血或促癌细胞转移的情况[45]。
4 问题与展望
综上所述,现将目前已被研究验证的与ENG存在相互作用的蛋白质汇总(见Table 1)。基于目前的研究,可以暂时描绘出如下的ENG调控血管生成的机制:在血管生成活跃阶段,内皮细胞表达大量的ENG,致使血管内皮钙黏连蛋白介导细胞间黏附作用被削弱;同时,大量的ENG开始加速招募TGF-β家族蛋白质结合到细胞膜上的ALK1-ENG复合物,而fibronectin-α5β1复合物与ENG的互作又进一步加速了这一复合物的形成,导致配体与ALK1-ENG复合物的结合被进一步加强。随后,ENG被磷酸化,SMAD1/5信号被激活,内皮细胞开始分泌VEGF等因子;同时ENG又进一步延长了VEGF-A信号,致使内皮细胞的迁移与增殖活动被加强。此外,在GIPC的作用下,上述的各个信号作用时间又随着ENG驻膜时间的延长也被进一步延长,最终表现为活跃的血管出芽与血管生成等生理活动。但随着血管生成趋于成熟,已被磷酸化的ENG会随着β-arrestin2或被磷酸化的ALK1一同被胞吞,致使ENG在细胞膜上介导的ALK1-SMAD1/5通路被抑制。与此同时,内化的ENG又开始抑制ERK信号。此外,随着ENG被一同胞吞的α5β1会同时激活FAK信号,最终导致内皮细胞的分裂与迁移活动均被抑制,表现为内皮细胞趋于静默,血管最终成熟。在血管衰老阶段,内皮细胞开始大量表达S-ENG,ALK5-SMAD2/3信号被增强,内皮细胞开始大量表达CTGF等促进细胞外基质形成的因子,可能进一步加速血管的衰老。

Table 1 The proteins interacted with ENG
因此,ENG通过调控信号通路和介导细胞间相互作用,在血管生成中发挥着重要作用。但ENG是如何在错综复杂的细胞环境中协调各信号的有序传导,仍有待研究。ENG作为调控血管生成的重要蛋白质,是一个具有药物研发前景的靶蛋白质。虽然目前的ENG单克隆抗体TRC105在靶向治疗肿瘤方面初见成效,但仍因为抗药性和适用范围小而收效甚微。在未来的研究中,解析ENG与不同蛋白质互作结构,深入理解ENG与不同蛋白质的互作机制,进而对不同病患实施不同的鸡尾酒疗法,或许可以对血管类疾病的治疗提供更有针对性的帮助。
