小胶质细胞对视网膜及神经系统疾病的监控作用
2022-09-07许佳综述金子兵审校
许佳 综述 金子兵 审校
首都医科大学附属北京同仁医院 北京市眼科研究所,北京 100005
小胶质细胞是中枢神经系统(central nervous system,CNS)定植的巨噬细胞。生理情况下,小胶质细胞通过伸缩的分枝监测视网膜及神经系统微环境,并通过突触修剪调节突触发育和可塑性,对视网膜和神经系统发育及稳态有重要作用;病理条件下,小胶质细胞对短暂损伤能迅速做出激活、迁移和吞噬活性增加反应,有免疫保护作用[1-2]。然而,在持续的病理刺激下,小胶质细胞炎症反应失调,使疾病恶化。小胶质细胞在视网膜及神经系统疾病中发挥免疫保护和神经毒性的双重作用,极易过度活化造成自身损伤,目前其调控视网膜及神经系统免疫稳态的作用机制尚不清楚。本综述对小胶质细胞在视网膜疾病和涉及眼部的神经系统疾病的研究进展及待解决问题进行梳理,希望对深入研究相关疾病的发病机制及治疗提供帮助。
1 小胶质细胞的历史发现
1856年,德国病理学家Rudolf Virchow用glia来描述整个CNS的非神经元腔室,其细胞成分未知,直到1919年西班牙神经学家Pio del Rio Hortega发现其中有一群单独的细胞类型,并称之为小胶质细胞(microglia)[1-2]。
2 小胶质细胞的发育来源
小胶质细胞发育来源尚存在争议。研究发现,小鼠脑内小胶质细胞是由胚胎7.25 d(E7.25)左右卵黄囊中C-kitloCD41lo的骨髓祖细胞发育而来[1,3-4],发育过程离不开集落刺激因子1受体(colony stimulating factor-1 receptor,CSF-1R)、转录因子PU.1和IRF8[5-7](图1)。小鼠脑中可能潜伏着小胶质细胞的前体细胞,可在一定程度上补充减少的小胶质细胞[8]。小鼠出生后第1个月是神经元及突触发育的关键阶段,小胶质细胞通过分泌神经营养因子如脑源神经营养因子(brain derived neurotrophin factor,BDNF)影响突触可塑性,进而影响学习和记忆,通过吞噬功能促进神经元存活、突触修剪及突触可塑性,促进神经系统的发育[9-10]。
3 小胶质细胞的生物学功能
静息状态的小胶质细胞胞体很小,呈短棒状,伸出数支条状突起,突起表面粗糙,有棘刺;胞核较小,约5 μm,形态不规则,可呈肾形、椭圆形或三角形,核染色质多;激活的小胶质细胞体积明显增大,胞体呈现阿米巴样改变[11]。小胶质细胞与神经系统其他细胞相互作用,在维持大脑稳态和组织损伤修复等病理状态具有不同的形态和功能[12]。
小胶质细胞的激活因素为:(1)代谢相关因素 如细胞外钾离子浓度增高、脂类、淀粉样肽、钙调基因相关肽、缺血、缺氧、脑缺血时的高血糖;(2)细胞因子类 干扰素和白细胞介素(interleukin,IL)-4可降低其吞噬能力;IL-6和肿瘤坏死因子(tumor necrosis factor,TNF)-α可促进其功能;IL-34对小胶质细胞发挥功能有重要作用;(3)外源性物质 细菌、病毒、生物蛋白、重金属等[13-14]。激活的小胶质细胞一方面通过释放和分泌一系列免疫应答分子、细胞因子、神经毒性物质等产生细胞毒性效应;另一方面通过吞噬清除病原体和细胞碎片以及分泌抗炎物质、神经生长因子等,保护神经元的存活[15](图1,2)。
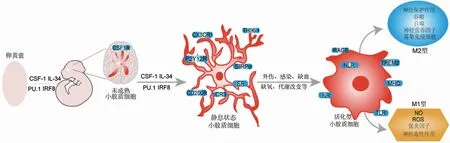
图1 小胶质细胞发育及功能总结 小胶质细胞上的分子分别代表CSF1R、THIK-1、SR、CR3、CX3CR1、P2Y12R、CD200R、NR、SIRPα、TLR、MHC、RAGE、NLR、TREM2 CSF:集落刺激因子;IL:白细胞介素
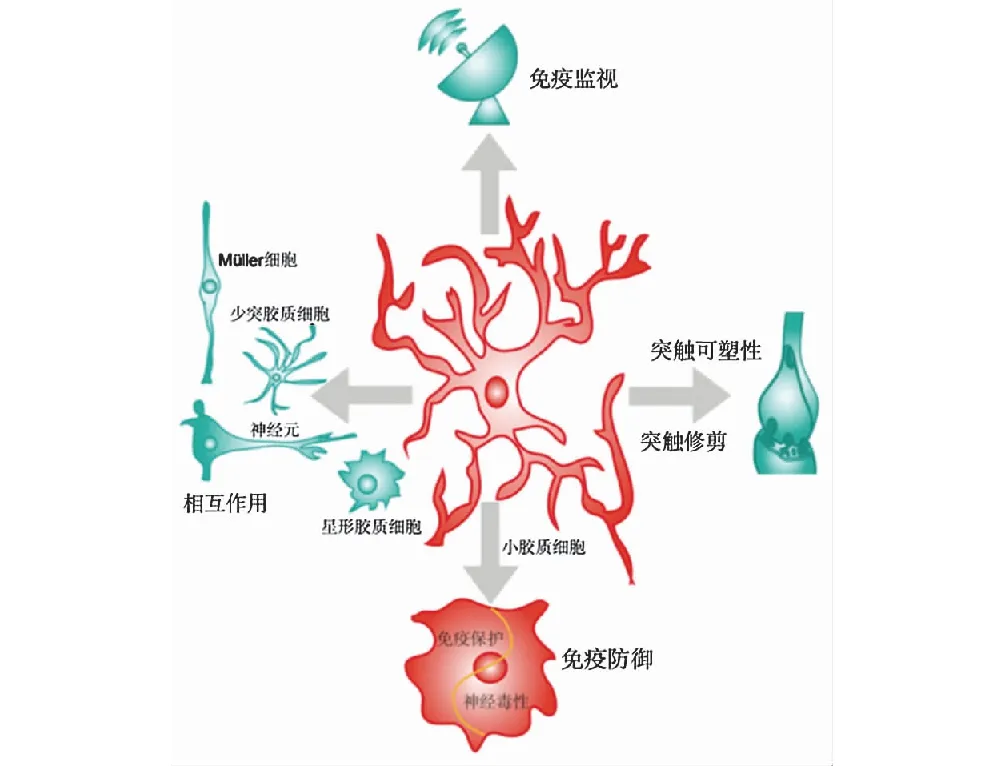
图2 小胶质细胞功能示意图
3.1 神经毒性作用
小胶质细胞过度活化产生的氧化应激、细胞因子及兴奋性递质是神经毒性的主要原因。小鼠皮质撞击后4 d和7 d,还原型烟酰胺腺嘌呤二核苷酸磷酸氧化酶2(nicotinamide adenine dinucleotide phosphate oxidase 2,NOX2)的表达和核因子(nuclear factor,NF)-κB的激活主要发生在小胶质细胞中;抑制NOX2,可减少髓系细胞活性氧的产生,保护神经元免受氧化损伤;这些变化与脑损伤后小胶质细胞NF-κB通路明显下调以及促炎细胞因子TNF-α和IL-1β的产生减少有关[16]。脂多糖和干扰素激活的小胶质细胞通过干扰素调节因子-1诱导细胞毒性一氧化氮(nitric oxide,NO)产生,通过诱导半胱氨酸天冬氨酸蛋白酶11(cysteinyl aspartate specific proteinase 11,caspase-11)表达启动不依赖于NO的细胞凋亡途径[17];在小鼠发育的海马中,小胶质细胞表面的整合素蛋白CD11b和免疫原受体DAP12控制着小胶质细胞超氧化物离子的产生,从而诱导神经元死亡[18]。
3.2 神经保护作用
小胶质细胞的神经保护作用依赖其免疫细胞的本质,即抗原提呈功能、分泌功能、吞噬功能及与神经系统其他细胞相互作用等功能[2],这些功能的发挥离不开其受体的作用。
3.2.1免疫防御相关受体 (1)Toll样受体 Toll样受体(Toll-like receptor,TLR)是小胶质细胞表达的主要受体,其中TLR1、TLR2、TLR4、TLR6主要存在于细胞表面并作为识别病变部位的第一道防线,TLR3、TLR7和TLR9是细胞内识别病毒细菌核苷酸序列的受体[19];TLR活化后激活NF-κB、丝裂原激活蛋白激酶(mitogen activation protein kinase,MAPK)及干扰素等信号通路,促进IL-6、TNF-α、IL-23释放,引起炎症反应[1];此外,活化的小胶质细胞还能释放神经营养因子,如BDNF,胰岛素样生长因子(insulin-like growth factor,IGF)等发挥神经保护作用[20];(2)主要组织相容性复合体Ⅰ类和Ⅱ类 主要组织相容性复合体(major histocompatibility complex,MHC)-I类分子和MHC-Ⅱ类分子参与抗原提呈,启动免疫应答;(3)NOD样受体 NOD样受体(NOD-like receptor,NLR)是小胶质细胞内的一种模式识别受体系统。NLR家族成员NLRC4和NLRP3蛋白的信号转导通路可以控制caspase-1和NF-κB的激活,促进IL-1β和IL-18分泌,介导炎症反应[21];(4)补体受体 小胶质细胞表达补体受体(complement receptor,CR)。在神经退行性疾病,如阿尔兹海默病(Alzheimer disease,AD)中,慢性补体激活与突触和神经元丢失有关[22]。在健康的小鼠脑组织中,C3和C1q广泛表达在未成熟的突触中,小胶质细胞表达CR3,通过C3-CR3信号通路发挥吞噬作用,缺少C1q、C4、C3或CR3使突触连接损伤,这说明了脑组织的正常功能需要经典补体途径的激活;在许多CNS疾病中,补体蛋白在神经元丢失的迹象出现之前就显著上调,这表明补体介导的突触清除可能推动疾病进展[23]。补体激活在AD发病早期对疾病发展起抑制作用;研究表明小鼠中补体系统介导的突触重排会影响记忆功能[24];(5)CD200R CD200R是一种糖蛋白,小胶质细胞表面的CD200R介导其与神经系统其他细胞相互作用,在免疫反应中起抑制性作用,防止小胶质细胞过度活化,维持体内稳态[25];(6)晚期糖基化终末产物受体 晚期糖基化终末产物受体(advanced glycation end product receptor,RAGE)是小胶质细胞的多配体受体,可以识别β-淀粉样蛋白(amyloid β-protein,Aβ)、糖化蛋白和其他β纤维化的蛋白介导炎症反应[26];(7)清道夫受体 清道夫受体(scavenger receptor,SR)家族分为A型和B型,小胶质细胞上的SR参与微生物和纤维化Aβ的识别和吞噬,清除氧化的低密度脂蛋白及长链脂肪酸等,在免疫防御系统中起重要作用[27];小胶质细胞表面表达多种SR,包括CD36、CD68、Scarb1、Scarf、CXCL16和Scara1等。其中,CD36和Scara1可以结合Aβ,在Aβ清除过程中可能有作用。CD36属于B型SR,还是TLR4的辅助受体,小胶质细胞通过CD36吞噬胞外聚合的Aβ,激活NLRP3炎性小体,进一步激活Caspase1从而促进IL-1β和IL-18的成熟和释放[28]。Scara1是A型SR,AD小鼠脑中Scara1的表达水平随着年龄增长而下降,提示其可能影响淀粉样蛋白的沉积[29];(8)髓样细胞触发性受体2 髓样细胞触发性受体2(triggering receptor expressed on myeloid cells-2,TREM2)可与磷脂、硫脂、脂多糖和DNA等配体结合。TREM2通过与衔接蛋白TYROBP/DAP12结合介导机体对细菌等病原体以及凋亡或死亡的神经细胞、细胞碎片的吞噬和清除过程[30]。
3.2.2免疫监视相关受体 小胶质细胞多条cAMP调控的丝状伪足能够进行纳米级监测,是其发挥免疫监视的主要方式,可迅速活化发挥免疫保护功能[31];小胶质细胞的钾离子通道蛋白是介导免疫监视的重要分子,在小胶质细胞活化后帮助释放炎性因子IL-1β,保障神经系统的稳态[32]。
3.2.3细胞因子受体 CSF-1和IL-34均可结合CSF-1R,介导小胶质细胞分化发育,IL-34-CSF-1R通路起主导作用[6];小胶质细胞上趋化因子受体CX3CR1与趋化因子CX3CL1/fractalkine结合引发钙离子内流产生趋化反应,诱导细胞到生物体特定部位,缺少CX3CR1会导致IGF1产生减少,影响神经元存活[33]。
3.2.4突触可塑性相关受体 小胶质细胞表达谷氨酸离子型受体,如α-氨基-3-羟基-5-甲基-4-异恶唑丙酸受体(α-amino-3-hydroxy-5-methyl-4-isoxazole propionic acid receptor,AMPAR),N-甲基-D-天门冬氨酸(N-methyl-D-aspartate,NMDA)受体(NR)和谷氨酸代谢型受体,如mGlutR2,参与神经突触发育以及信号转导,过度兴奋也会导致神经毒性,AMPAR抑制TNF-α释放,而mGlutR2促进TNF-α释放介导神经毒性[20]。NR激活后引起钙离子内流,随后ATP释放介导兴奋性神经毒性在AD发病中发挥重要作用,NR过度激活,会损伤神经细胞[34]。NR1与钙离子通道开放密切相关,被认为是突触可塑性及海马和皮质神经元长时程增强(long-term potentiation,LTP)的主要调控者,而LTP的损害与AD密切相关[35-36]。
3.2.5突触修剪相关受体 小胶质细胞上的嘌呤能P2Y12受体(P2Y12R/P2RY12)在其活化后下调表达介导小胶质细胞的迁移、吞噬以及突触修剪[20];突触表面的CD47与小胶质细胞上的SIRPα结合作为“don't eat me”信号避免突触过度修剪[9];小胶质细胞自噬在神经系统突触形成及修剪过程中也发挥重要功能[37];小胶质细胞CX3CR1参与CNS及视网膜神经元的突触形成和突触修剪从而影响神经元的正常发育[38]。
4 小胶质细胞与神经系统疾病的联系
4.1 小胶质细胞与神经退行性眼病
小胶质细胞参与视网膜色素变性(retinitis pigmentosa,RP)和X连锁青少年视网膜劈裂症等遗传性视网膜疾病,以及年龄相关性黄斑变性、糖尿病视网膜病变、青光眼、葡萄膜炎、神经系统疾病等多因素视网膜疾病的发生和发展[39]。
RP是一种以感光细胞进行性退化为特征的疾病,是全世界年轻人无法治愈盲的重要原因。研究发现与正常小鼠相比,CX3CR1缺陷的rd10小鼠中,小胶质细胞向感光细胞层浸润显著增加,并与感光细胞凋亡和萎缩加速相关;小胶质细胞缺少CX3CR1与炎性因子和小胶质细胞活化标志物的表达增加有关,外源性CX3CL1玻璃体内注射增加rd10小鼠视网膜CX3CL1-CX3CR1信号,可显著减少小胶质细胞浸润、吞噬和活化,改善感光细胞的退化[40]。这些结果表明,CX3CL1-CX3CR1信号是调节小胶质细胞介导的神经退形性变的分子机制之一,是RP治疗的潜在靶点。研究发现RP患者和rd10小鼠模型视网膜中多种补体成分明显上调,这种改变与感光细胞变性一致,C3表达增加,活化小胶质细胞。C3的缺失加速了感光细胞结构和功能的退化,并改变了视网膜炎症基因的表达。CR3的基因缺失重现了这些表型,提示C3-CR3信号是介导小胶质细胞-感光细胞相互作用的调节因子。C3或CR3的缺乏降低了小胶质细胞对凋亡感光细胞的吞噬能力,增加了小胶质细胞对感光细胞的毒性,表明补体介导的小胶质细胞清除凋亡感光细胞在RP中具有新的适应性作用[41]。
在出血性黄斑变性小鼠模型中,用米诺环素治疗可以防止小胶质细胞在视网膜下空间聚集,增加感光细胞存活率[42];然而,在视网膜脱离(retinal detachment,RD)的情况下,小胶质细胞的消融阻止了其在视网膜下空间的聚集,降低感光细胞存活率[43]。RD是视网膜疾病中常见威胁视力的并发症,RD发生后12 h内迅速导致感光细胞死亡。小胶质细胞标志物P2ry12和小胶质细胞的耗竭证明小胶质细胞在RD后24 h内迅速激活并迁移到损伤区域,一旦进入受损的感光细胞层,就可以观察到激活的小胶质细胞在其细胞体内含有自体荧光,这表明其功能是吞噬受损或死亡的感光细胞。小胶质细胞耗竭导致疾病恶化和抑制巨噬细胞浸润,提示小胶质细胞参与调节视网膜的神经炎症,介导RD中感光细胞存活[43]。Todd等[44]研究发现,小胶质细胞在兴奋性毒性损伤的视网膜中提供神经保护,这可能是通过小胶质细胞产生的IL-1β与星形胶质细胞上的IL-1R1结合发挥作用。
青光眼是一种以视网膜神经节细胞(retinal ganglion cell,RGC)变性为特征的视神经退行性疾病,可造成不可逆转的视力损害和盲,其具体机制尚未完全阐明。目前,大量体内及体外研究发现,视网膜胶质细胞,如Müller胶质细胞、星形胶质细胞和小胶质细胞与青光眼的发生和发展,特别是RGC的损伤密切相关[45-46];在斑马鱼视网膜中,小胶质细胞和巨噬细胞缺乏会导致Müller细胞行为失调。因此,小胶质细胞和Müller细胞信号传导对于解锁Müller细胞的再生潜能以修复受损的视网膜至关重要[47]。
在青光眼小鼠模型中,补体的上调发生在RGC死亡之前,RGC的突触被小胶质细胞吞噬,抑制补体可以抑制青光眼小鼠RGC的退化[48]。DBA/2J小鼠是青光眼模型构建的常用小鼠,米诺环素是具有抗炎和抗凋亡特性的第2代四环素,研究发现米诺环素可减少小胶质细胞的活化,改善RGC轴突的运输和完整性,但对年龄相关的眼部特征性改变无影响,其机制可能为米诺环素增加了具有静止分枝形态的小胶质细胞比例,并降低了Iba1(与激活相关的小胶质细胞特异性钙配体)的mRNA和蛋白表达水平,并且小胶质细胞激活的减少与RGC轴突运输的显著改善相关联,表明在青光眼中,视网膜和视神经乳头的小胶质细胞活化可能是视神经功能早期下降及其随后退化的一个因素[49]。
青光眼中小胶质细胞激活后,炎性蛋白表达上调。在大鼠高眼压模型中使用TNF-α阻断剂依那西普后,发现即使眼压持续升高,视盘周围表达TNF-α的活化小胶质细胞显著减少,从而防止RGC的损伤和减少,这些发现说明小胶质细胞的过度激活加重了青光眼的神经元损伤,提出了一种使用TNF-a拮抗剂或炎症抑制剂治疗青光眼的新策略[50]。用经角膜缘激光光凝模型诱导大鼠眼压升高模型检测米诺环素的作用及机制,发现米诺环素能显著增加细胞的抗凋亡能力,使抗凋亡基因Bcl-2表达增加,减少IL-18的表达及青光眼中活化的小胶质细胞数量,从而使青光眼病情好转[51]。
视网膜中小胶质细胞主要分布在内丛状层和外丛状层,敲除小鼠IL-34基因后,内丛状层小胶质细胞数量显著减少,视网膜电流降低,而外丛状层没有发生变化,这表明IL-34对小胶质细胞的维持作用仅发生在内丛状层,具有区域特异性;进一步在视网膜变性模型中研究发现,小胶质细胞能够迁移到视网膜外核层保护视网膜色素上皮细胞。由此可见,不同情况下视网膜中的小胶质细胞可能发挥不同的作用,对视网膜小胶质细胞的功能及机制研究将有助于视网膜退行性疾病的治疗[52]。
因此,小胶质细胞在视网膜中的功能具有两面性,如何控制小胶质细胞的功能使其有利于视网膜的存活,减轻神经毒性及炎症对神经退行性眼病的治疗具有重要意义。
4.2 小胶质细胞与帕金森病
帕金森病(Parkinson disease,PD)是一种神经退行性运动障碍,表现为黑质中多巴胺能神经元丢失,伴随着由α-突触核蛋白(α-synuclein,α-SYN)聚集体组成的路易体小体,涉及视网膜多巴胺能细胞变性[53]。PD患者小胶质细胞中TLR2表达水平升高,α-SYN激活TLR1/2后以MyD88依赖方式释放促炎细胞因子TNF-α和IL-1β;另外,激活的小胶质细胞也释放抗炎细胞因子,提示小胶质细胞TLR1/2是α-SYN的受体,通过释放细胞因子参与PD进展[53]。在PD小鼠模型中,缺乏CX3CR1的小鼠表现出α-SYN介导的炎症反应和小胶质细胞吞噬功能降低,说明CX3CL1-CX3CR1信号在PD中的重要性[54]。PD中M1样小胶质细胞释放的促炎因子常伴随着多巴胺能神经元的丢失;相反,M2样活化的小胶质细胞分泌抗炎因子IL-4、IL-13、IL-10、TGF-β和IGF1减轻炎症,加速修复[15,20,52,55]。
4.3 小胶质细胞与AD
AD是一种以进行性认知障碍和记忆力损害为主要表现的CNS退行性病变。该疾病的标志是Aβ的细胞外沉积和高磷酸化tau蛋白(phospho Tau protein,pTau)在神经内积聚,这些沉积物还存在于视网膜中[30]。小胶质细胞对Aβ积聚和神经退行性损伤有反应,进化为疾病相关小胶质细胞引起的炎症反应和神经毒性是AD核心病理机制[20,30]。
载脂蛋白E(apolipoprotein E,ApoE)的等位基因ε4是晚期AD的危险因素。通过全基因组关联分析发现与AD发病显著相关的易感位点聚集素和磷脂酰肌醇结合型胞苷组装蛋白;进一步分析发现补体受体、桥联整合子1基因和跨膜4A基因在AD患者中突变增多[56]。还有研究报道TREM2、磷脂酰肌醇磷脂酶Cγ2和CD33的单核苷酸多态性也与晚发性AD的风险增加有关[57]。携带TREM2突变R47H的个体患AD风险显著增加,TREM2-DAP12-DAP10信号触发蛋白质和脂质磷酸化级联反应,导致钙离子动员、整合素活化、细胞骨架重排以及能量代谢激活[30],该信号通路可能与AD的发生有关。TREM2-APOE通路介导神经退行性小胶质细胞表型的转变,提示其可以作为新的靶点,帮助小胶质细胞恢复稳态[58]。用AD小鼠模型进行研究发现泛素连接酶COP1通过蛋白酶体途径降解小胶质细胞中的转录因子c/EBPβ来抑制神经炎症,防止小胶质细胞过度激活,影响小胶质细胞的激活状态,减轻神经退行性病变,减少pTau蛋白水平,揭示了AD疾病过程中小胶质细胞基因表达的新机制,为疾病治疗提供潜在的作用靶点[59]。综上,寻找限制小胶质细胞产生神经毒性物质同时不削弱其吞噬作用的药物或方法可能成为延迟神经变性疾病发生的新策略。
4.4 小胶质细胞与多发性硬化
多发性硬化(multiple sclerosis,MS)是一种CNS脱髓鞘性自身免疫性疾病,病变位于脑、视网膜神经节和骨髓,病灶播散广泛可伴有神经退行性病变。TMEM119是MS中小胶质细胞的表面标志物[60]。小胶质细胞的吞噬受体Mer受体酪氨酸激酶突变会增加MS的发生率[61];Werneburg等[62]利用尸检MS组织、临床前非人灵长类MS模型和2种脱髓鞘疾病的啮齿动物模型,研究视觉系统中突触的变化,发现小胶质细胞介导突触吞噬和突触丢失。突触丢失可能发生在MS相关病理改变的早期或之前,并与补体C3相关。
综上,小胶质细胞在多种神经系统疾病的发生和发展过程中起重要作用(表1),精细调控小胶质细胞的神经保护作用和神经毒性作用有助于神经系统疾病的治疗。

表1 小胶质细胞生理功能及疾病情况下相关的基因及通路正常功能及疾病基因/通路参考文献突触修剪CR3,CX3CR1[9,15]突触可塑性TLR4,CX3CR1[1,15]神经元死亡CR3,AXL,MER,DAP12[15]神经退行性眼病DAP12,CX3CR1,CR[43,52]ADTREM2,DAP12,APOE,CD33,CR1,PLCG2,ABCA7,BIN1[56]PDTREM2,TLR1/2,Notch信号通路[53,55]MSTLR4,MYD88,MER[60-62] 注:AD:阿尔兹海默病;PD:帕金森病;MS:多发性硬化
5 展望
小胶质细胞是神经系统的忠诚卫士,可识别“自我”监视着“非己”,第一时间对外界变化作出免疫应答。从细胞分子水平上揭示其在神经系统中的激活过程及其作用机制可能为开发治疗神经系统疾病有效药物提供潜在的靶点。虽然小胶质细胞在不同疾病中的作用受到较大关注,但其在生理情况下的正常功能还不清楚,在视网膜及神经系统疾病中的作用也需要进一步阐明:(1)小胶质细胞是如何进行免疫监视维持动态平衡的?是否有区域特异性?睡眠状态和清醒状态下小胶质细胞的免疫监视功能有何不同?(2)决定小胶质细胞发挥损伤作用还是保护作用是由哪些信号通路介导的?控制平衡的关键点是什么?如何减少有害作用?(3)小胶质细胞与神经系统其他细胞相互作用的方式有何差别?(4)小胶质细胞的自噬和死亡如何调控?向周围传递什么信号?(5)眼作为重要的视觉神经中枢器官,与大脑的信号传导密切相关,眼不仅可以看物体,还具有传达情绪、记忆、应激反应等多种属性,那么视网膜中小胶质细胞有哪些独特功能和作用机制?(6)人和小鼠的小胶质细胞有何异同?这一系列问题都有待进一步研究,从而合理利用其保护功能,减少有害作用,为预防和治疗CNS疾病提供潜在靶点。人视网膜及神经组织小胶质细胞难以获取从而使人小胶质细胞的研究变得困难。但随着类器官,尤其是视网膜类器官、光遗传学、表观遗传学、单细胞测序、质谱流式及成像等科学理论和技术的发展,对这些问题的深入研究将为理解小胶质细胞在正常情况下及神经系统疾病中究竟扮演何种角色提供重要启示,并将有助于最终找到针对神经系统疾病的新的药物治疗靶点,有助于疾病的精准治疗。
利益冲突所有作者均声明不存在利益冲突
