利用菱镁矿制备非晶态硅酸镁
2022-09-06王琪浩王余莲林永瑾朱益斌刘珈伊时天骄田伊笛苏德生袁志刚
王琪浩,王余莲,薛 铭,林永瑾,朱益斌,张 俊,刘珈伊,时天骄,田伊笛,李 闯,苏德生,袁志刚
(1.沈阳理工大学 材料科学与工程学院,辽宁 沈阳 110159;2.东北大学 资源与土木工程学院,辽宁 沈阳 110819; 3.辽宁省超高功率石墨电极材料专业技术创新中心,辽宁 丹东 118000)
据统计,我国菱镁矿储量约占全世界总储量的2/3,其中辽宁省菱镁矿资源储量最丰富,占全国总储量的85.62%[1]。基于我国菱镁矿资源储量丰富这一资源优势,利用菱镁矿制备硅酸镁,具有原料廉价易得、来源广泛等优点。
人工合成硅酸镁(xMgO·ySiO2·nH2O)是一种具有高比表面积、大孔容积、类似海泡石2∶1型链状和层状过渡型结构的材料,因空穴和孔道丰富,具有良好的吸附性能,被广泛应用于废水处理和油污吸附领域[2]。除此之外,硅酸镁在保温阻燃[3]、食品加工[4]、医学[5]等领域具有广阔的应用价值。
目前,人工合成硅酸镁主要方法有沉淀法、水热法和模板法。高慧慧等[6]以稻壳为原料,热处理得到无定形二氧化硅(SiO2)溶液,再将镁离子(Mg2+)、尿素、质量浓度为1.67 g/L无定形二氧化硅溶液混合,获得花状型硅酸镁;李传亮等[7]以高低温盐、泡花碱为原料,制备达到《中国药典》二部标准要求的医药级三硅酸镁;冯凌等[8]通过共沉淀法将硅酸钠(Na2O·nSiO2)、硝酸镁(Mg(NO3)2)混合制备大比表面积的多孔非晶态三硅酸镁粉体;张丽丽[9]通过硅胶负载、溶胶-凝胶、水热等方法制备了一系列性能优良的硅酸镁;Wang等[10]将坡缕石、浓度为0.915 mol/L硅酸盐溶液、浓度为0.686 mol/L镁盐溶液混合,通过水热法制备出比表面积为407.3 m2/g的硅酸镁;Clowutimo等[11]通过水热法,将硅胶球、去离子水、氯化铵(NH4Cl)、氨水(NH3·H2O)、六水氯化镁(MgCl2·6H2O)混合,经过沉淀、分离、烘干后得到球型多孔硅酸镁。
方娴韵等[12]以纳米SiO2为模板、以氯化镁(MgCl2)为镁源,在弱碱性条件下制备了纳米空心球状硅酸镁;Yang等[13]以空心管状四氧化三铁-二氧化硅-银(Fe3O4-SiO2-Ag)为模板、硫酸镁(MgSO4)为镁源、氨水为沉淀剂,通过模板法制备比表面积为539.36 m2/g、对亚甲基蓝有较好吸附能力的磁性空心纳米管状硅酸镁;Lu等[14]采用介孔模板辅助水热法制备比孔体积为0.84 cm3/g、比表面积为446 m2/g、对亚甲基蓝的吸附容量为418 mg/g的介孔硅酸镁材料;毛丽莉等[15]将硅酸镁颗粒、聚偏氟乙烯的N-N二甲基乙酰胺溶液充分混合后,通过挤出成型、烘干获得硅酸镁吸附剂。
综上,目前硅酸镁的制备多以MgSO4[16]、Mg(NO3)2[17]或MgCl2[18-19]等化学试剂为原料,存在成本较高、废液处理困难的问题。本文中以菱镁矿为原料,经过煅烧、水化、碳酸化等方法制备所得重镁水为镁源,五水合硅酸钠(Na2SiO3·5H2O)为硅源,通过沉淀反应制得硅酸镁,主要研究镁、硅物质的量比,体系pH,反应时间,煅烧时间等因素对硅酸镁制备的影响。
1 实验
1.1 试剂、材料和仪器设备
Na2SiO3·5H2O,分析纯,纯度为99.8%(质量分数,下同),天津市大茂化学试剂厂;重镁水,利用菱镁矿为原料,通过煅烧、水化、碳酸化法自制;二氧化碳(CO2),抚顺嘉和气体有限公司;去离子水,自制。
DF-101S型集热式恒温加热磁力搅拌器(巩义市予华仪器责任有限公司);1400-40型HMF马弗炉(上海皓越电炉技术公司);JJ-1型精密定时电动搅拌器(常州荣华仪器制造有限公司);FA2004B型精密天平(北京赛多利斯仪器有限公司);DHG-9140B型电热恒温鼓风干燥箱(中仪国科(北京)科技有限公司)。
1.2 样品制备
1.2.1 重镁水制备
称取一定质量的轻烧镁粉(煅烧菱镁矿所得)与去离子水按质量比1∶40混合。将混合液于60 ℃水化3.5 h,获得水化溶液;往水化溶液中通入CO2并于冰水水浴下搅拌,进行碳酸化,待溶液pH为7.5时,停止通气;搅拌、抽滤,得到澄清滤液为重镁水,即Mg(HCO3)2溶液,浓度为0.3 mol/L。
1.2.2 硅酸镁制备
称取一定量五水合硅酸钠,配置浓度为1 mol/L硅酸钠溶液。将不同体积(18~54 mL)的硅酸钠溶液以不同滴加时间(15~65 min)加入60 mL浓度为0.3 mol/L的重镁水中,并在温度为25 ℃条件下恒温搅拌,得到混合溶液。向混合溶液中通入CO2将溶液pH调节至8.5~10.5。反应结束,经抽滤、洗涤、干燥、研磨得到样品。将样品在温度为400 ℃条件下煅烧一定时间(0~4 h)获得白色粉末。
1.3 测试与表征
利用X射线衍射仪分析产物物相组成,扫描角度5°~90°,扫描速率10 (°)/min;采用扫描电子显微镜观察产物形貌;利用傅里叶变换红外光谱仪测定产物基团;借助全自动比表面分析仪测定产物比表面积。
2 结果与讨论
2.1 镁、硅物质的量比对产物比表面积的影响
固定重镁水与硅酸钠溶液混合体系的pH为10,滴加硅酸钠溶液时间为65 min,煅烧时间为4 h,探究镁、硅物质的量比(1∶1、1∶1.5、1∶2、1∶2.5、1∶3)对产物物相组成和比表面积的影响,结果如图1、2所示。

图1 镁、硅物质的量比不同时所得产物的XRD图谱Fig.1 XRD patterns of products obtained at different mole ratio of magnesium to silicon
由图1可知,不同镁、硅物质的量比所得产物在2θ=29.2°、35.5°、60.2°处宽化的馒头形特征峰均与羟基硅酸镁(Mg3Si2O5(OH)4,JCPDS22-1162)[20-21]标准峰对应,表明所得产物为非晶态羟基硅酸镁。由此说明以天然菱镁矿为镁源,可以制备硅酸镁,但结晶度较差,且镁、硅物质的量比降低对产物物相组成和结晶度没有明显影响。
由图2可知,随着镁、硅物质的量比减小,产物比表面积先增大后减小。镁、硅物质的量比为1∶1.5时,比表面积达最大值167.25 m2/g。镁、硅物质的量比继续减小,比表面积减小,这可能是由于初始阶段硅酸钠的加入促进反应进行;当过量硅酸钠加入至溶液中,易生成硅酸胶体。故选择1∶1.5为适宜的镁、硅物质的量比。

图2 镁、硅物质的量比不同时所得产物的比表面积变化曲线Fig.2 Specific surface area curve of products obtained at different mole ratio of magnesium to silicon
2.2 混合体系pH对产物比表面积的影响
固定滴加硅酸钠溶液时间为65 min,镁、硅物质的量比为1∶1.5,煅烧时间为4 h,探究体系pH=8.5、9.0、9.5、10.0、10.5对产物物相组成以及比表面积的影响,结果分别如图3、4所示。
从图3可以看出,混合体系pH不同时所得产物在2θ=29.2°、35.5°、60.2°处弥散的特征峰均与羟基硅酸镁(Mg3Si2O5(OH)4,JCPDS22-1162)标准峰一致,表明所得产物为非晶态羟基硅酸镁,但衍射峰趋于尖锐,表面碱性环境有利于提高硅酸镁结晶度。

图3 pH不同时所得产物的XRD图谱Fig.3 XRD patterns of products obtained at different pH
由图4可知,随着pH增大,比表面积先增大后减小,当pH为10,比表面积达最大值167.25 m2/g。随着pH继续增大,溶液中OH-数量增加,硅酸镁形成层状结构,羟基在碱性条件下稳定性较高,故比表面积增大;pH为10时,溶液碱性过强,破坏硅酸镁水解平衡,镁离子析出,层状结构破坏,比表面积减小。故选择10为适宜的pH。

图4 pH不同时所得产物的比表面积变化曲线Fig.4 Specific surface area curve of products obtained at different pH
2.3 反应时间对产物比表面积的影响
固定重镁水与硅酸钠溶液混合体系的pH为10,镁、硅物质的量比为1∶1.5,煅烧时间为4 h,探究硅酸钠反应时间(0、15、30、50、65 min)对产物物相组成及比表面积的影响,结果见图5、6。
由图5可以看出,所得产物在2θ=29.2°、35.5°、60.2°处宽化的馒头形特征峰均与羟基硅酸镁(Mg3Si2O5(OH)4,JCPDS 22-1162)标准特征峰对应,表明所得产物为非晶态羟基硅酸镁,且可以观察到,随着反应时间延长,2θ=35.5°、60.2°处的特征峰峰强减弱。
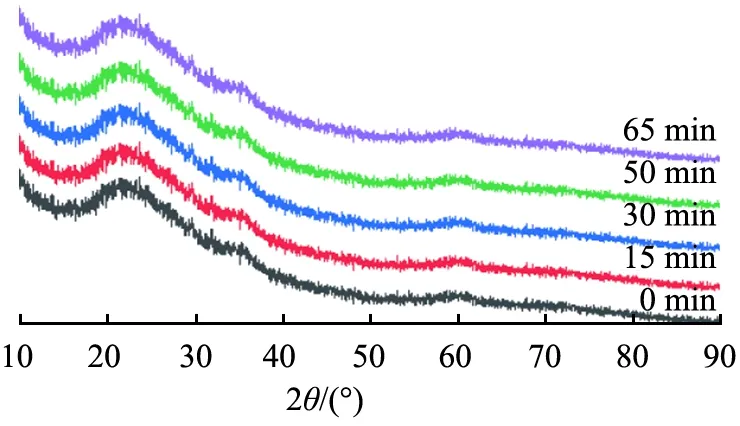
图5 反应时间不同时所得产物的XRD图谱Fig.5 XRD patterns of products obtained at different reaction time
由图6可以看出,随着反应时间延长,比表面积先减小后增大,当滴加时间为65 min时,比表面积达最大值84.4 m2/g。原因可能是滴加时间越长,硅酸钠溶液与重镁水反应越充分,晶核形成和晶粒成长较好,从而具有较大的比表面积。故选择65 min为适宜的滴加时间。
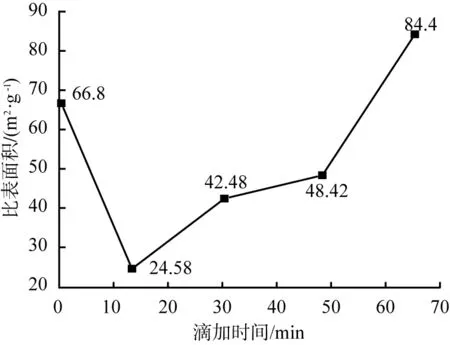
图6 反应时间不同时所得产物的比表面积变化曲线Fig.6 Specific surface area curve of products obtained at different reaction time
2.4 煅烧时间对产物的影响
固定重镁水与硅酸钠溶液混合体系的pH为10,滴加硅酸钠溶液时间为65 min,镁、硅物质的量比为1∶1.5,探究煅烧时间(0、1、2、3、4 h)对产物物相组成以及比表面积的影响,结果如图7、8所示。

图7 煅烧时间不同时所得产物的XRD图谱Fig.7 XRD patterns of products obtained with different calcination time
由图7可知,不同煅烧时间所得产物在2θ=29.2°、35.5°、60.2°处宽化的特征峰,均与羟基硅酸镁(Mg3Si2O5(OH)4,JCPDS22-1162)标准峰对应,表明所得产物为羟基硅酸镁。
由图8可知,未经煅烧处理时,硅酸镁比表面积最大,为44.18 m2/g;随着煅烧时间延长,硅酸镁比表面积逐渐减小,其原因可能是长时间煅烧导致样品中结晶水脱去,致使硅酸镁结构被破坏,层状结构消失,故比表面积减小,因此,选择不煅烧硅酸镁。

图8 煅烧时间不同时所得产物的比表面积变化曲线Fig.8 Specific surface area curve of products obtained with different calcination time
2.5 红外光谱与SEM分析
综上所述:利用菱镁矿制备硅酸镁的适宜条件为:重镁水与硅酸钠溶液混合体系pH为10,硅酸钠滴加时间为65 min,镁、硅物质的量比为1∶1.5,煅烧时间为0 h(不煅烧)。
图9为适宜条件所得产物的红外光谱图。由图可以看出,所得产物在波数为3 593、1 690、1 300、852、699、606 cm-1处附近出现吸收峰。3 593 cm-1处的吸收峰可归因于样品表面吸附水中的O—H基团;1 690 cm-1处吸收峰为O—H弯曲振动;1 300 cm-1处的吸收峰对应于产物中Si—O—Mg基团;852、699 cm-1处2个吸收峰则对应产物中Si—O—Si基团的弯曲振动和伸缩振动;而606 cm-1处的吸收峰则与Mg—O—H的伸缩振动和弯曲振动有关。由前述XRD分析结果可知,产物中含有Si、O、Mg、H元素,与产物FTIR光谱特征带基团相一致,进一步确定所得产物为硅酸镁Mg3Si2O5(OH)4。

图9 适宜条件所得产物的红外光谱图Fig.9 FTIR spectra of products obtained under suitable conditions
图10为适宜条件制备所得硅酸镁SEM图像。从图10(a)可以发现,沉淀法制得非晶态硅酸镁为不规则块状和颗粒状,其粒径为20 ~50 μm;从图10(b)可以发现,硅酸镁表面存在条状、颗粒状凸起和部分气孔状结构。

3 结论
1)以天然菱镁矿制备所得重镁水为镁源,采用沉淀法,当镁、硅物质的量比为1∶1.5,混合体系pH为10,反应时间为65 min,未煅烧,可制备比表面积为167.25 m2/g的不规则块状、颗粒状非晶态硅酸镁。
2)镁、硅物质的量比、混合体系pH、反应时间和煅烧时间对硅酸镁的物相组成的影响较小,但反应时间影响硅酸镁结晶度。镁、硅物质的量比与混合体系pH对硅酸镁比表面积影响较大;镁、硅物质的量比为1∶1.5相对于镁、硅物质的量比为1∶3的产物比表面积增加了79.67%;混合体系pH为10相对于混合体系pH为9.5的产物比表面积增加了83.19%。
