碳点与铋基氟化物纳米材料室温复合研究
2022-09-01朱婕禹庭盛昊阳陈叶青饶朋朋倪宗铭吴晓仪全志鹏
朱婕,禹庭,盛昊阳,陈叶青,饶朋朋,倪宗铭,吴晓仪,全志鹏
(五邑大学应用物理与材料学院, 广东江门 529020)
碳点(Carbon dots,CDs)作为一种最有前途的碳基纳米材料,其具有优异的发光性能[1]、可调谐发射[2]、低毒性[3-4]、易制备[5-7]等特点,近年来在生物成像、光催化、传感器和光电器件等领域引起了广泛的关注。与传统的光致发光材料相比,该材料的制备更加环保。然而,大多数情况下CDs 只有在溶液状态下才表现出明显的荧光性质,在聚集态或固态中荧光会发生猝灭,荧光猝灭归因于聚集状态下粒子间相互作用引起的非辐射跃迁。此外,由于CDs 表面含有丰富的亲水性官能团,导致固体CDs 通常难以获得,导致其在固态发光领域中的应用受到限制。克服荧光猝灭的通常方法是在固体基质(如聚合物、无机盐、二氧化硅、层状材料和介孔材料[8-14])中掺杂CDs。Qu 等[15]以聚乙烯醇(PVA)为基体的固态发射CDs 的量子效率(PLQY)为84%,而负载浓度仅为0.6%,表明较高的负载浓度会导致PLQY 的大幅下降,这主要是由于制备过程中固体局部聚集所致。此外,这些常见的方法伴随着多步骤的过程,不仅耗时而且伴随有不稳定的现象。此外,Qu 团队[16]还在泡沫结构中制备了尺寸均匀的CDs,其均匀结构带来了均匀的能带隙,显著抑制了聚集体中CDs之间的能量传递,从而克服了聚集体引起的猝灭,性能得到了很大的提高,但这种合成方法需要抽真空且反应时间相对较长和反应温度较高,这将不必要地增加合成成本。目前,大多数研究是先制备出长波长发射的CDs 溶液,然后再选择合适的基质,从而实现固态发光,这样的研究方法比较依赖于CDs本身的发光,严重影响CDs 在发光领域的应用发展。所以,迫切需要研发一种新途径来减弱对CDs发光依赖的同时而实现长波长固态发光。通过研究[17-18]发现,K0.3Bi0.7F2.4是一种合适的基质材料,它不仅成本低,而且在高温下热力学性质稳定。到目前为止,K0.3Bi0.7F2.4纳米颗粒作为铋基宿主体实现Ln3+掺杂的报道很少,大部分研究集中在上转换性能方面,而对其光致发光特性及其在白光发光二极管应用中的可行性研究不多。基于此,提出了一种通过室温共沉淀法制备CDs 基荧光粉的方法。选择柠檬酸(CA)为碳源、乙醇胺(EA)为氮源,采用水热法合成青光碳点溶液(C-CDs),然后其与K0.3Bi0.7F2.4基质合成C-CDs@K0.3Bi0.7F2.4纳米复合材料,该方法简单高效、无需高温高压环境,可在室温下快速合成。
1 实验部分
1.1 实验原料
柠檬酸(CA,99.5%)购买于阿拉丁公司(中国上海),乙醇胺(EA,99%)购买于麦克林公司(中国上 海)。分 析 级 的 五 水 硝 酸 铋Bi(NO3)3·5H2O(99%)和氟化钾(KF)试剂购自阿拉丁公司(中国上海),乙二醇(EG)是从北京化学试剂公司获得,氟化铵(NH4F,98%)产自国药化学试剂有限公司。所有的化学品都是直接使用的,没有进一步的纯化。
1.2 青色碳点(C⁃CDs)的制备
将0.630 4 g 的柠檬酸溶于30 mL 的去离子水中,然后加入3.621 mL 的乙醇胺,密封,在50 mL 的特氟龙内衬不锈钢高压釜中经8 h 加热至160 ℃,待反应结束后冷却至室温。将所得产物转移到离心管中,以10 000 r·min−1的转速离心10 min,除去未反应的颗粒。最后通过重新溶解在水中得到C-CDs溶液,以供进一步测量或使用。
1.3 C⁃CDs@K0.3Bi0.7F2.4复合材料的制备
首 先 将1 mmol 的Bi(NO3)3·5H2O、1 mmol 的KF 与10 mL 的EG 混合,待完 全溶解后加入1 mL 浓度为0.09 g·L−1的青色碳点溶液(C-CDs)。然后将6 mmol 的NH4F 溶解于20 mL 的EG 中。最后将两种溶液在室温下剧烈搅拌,再以1000 r·min−1速率离心,用无水乙醇超声洗涤2 次,在80 ℃/12 h 干燥,最终得到C-CDs@K0.3Bi0.7F2.4纳米复合材料。图1 为C-CDs@K0.3Bi0.7F2.4合成示意图。
图1 C-CDs@K0.3Bi0.7F2.4纳米复合材料的合成示意图Figure 1 Schematic diagram of the synthesis of C-CDs@K0.3Bi0.7F2.4 composites
1.4 表征手段
所有样品的XRD 测量在粉末衍射仪(X"Pert PRO,Cu Kα,λ=1.5418 Å)上进行,用扫描电子显微镜(SEM,JSM-6700F)对样品的形貌进行表征。使用JEOL JEM 2100 记录透射电镜(TEM)和高分辨率透射电镜(HRTEM)对样品的漫反射(DR)进行测量,用紫外-可见-近红外分光光度计(Lambda 950,Perkin Elmer)测量样品的吸收光谱,以BaSO4为标准参比。利用Aicolex Nexus 470 型红外光谱仪,通过混合样品与片剂KBr 获得红外光谱。以450 W 氙灯为光源,在爱丁堡仪器公司生产的FLS980 荧光光谱仪上对样品进行稳态和动态光谱分析。对于温度相关的样品的发射测量,在Linkam THMS600 冷热台上进行温度控制加热。X 射线光电子能谱(XPS)在Thermo Scientific K-Alpha 上进行分析。
2 结果与分析
2.1 C⁃CDs 及C⁃CDs@K0.3Bi0.7F2.4 纳米复合材料的光学性能
图2 为C-CDs 和C-CDs@K0.3Bi0.7F2.4的光 学 性能图。从C-CDs 的紫外-可见吸收光谱图可见,分别位于265 和355 nm 处有两个明显的吸收波段,前者的吸收带是由C═C 键的π—π*跃迁引起的,后者的吸收带是由C═O 键的n—π*跃迁引起的[19-20]。从C-CDs 激发-发射光谱可见,随着激发波长从400 nm增加到460 nm 时,C-CDs 的发射峰从477 nm(蓝色发射)红移到525 nm(绿色发射),并且发射强度逐渐减弱,在λex=420 nm 时达到最大值。值得注意的是,C-CDs 随着激发波长从400—460 nm 的变化,其发射强度和发射峰位都发生了变化,这与颗粒大小的变化及表面发射陷阱位置的分布有关。从C-CDs@K0.3Bi0.7F2.4漫反射光谱图可见,在深紫外区(200—320 nm)、近紫外区(320—420 nm)和蓝绿色区(450—580 nm)有三条宽吸收带,与C-CDs 不同的是,C-CDs@K0.3Bi0.7F2.4纳米复合材料在420—580 nm 范围内出现了明显的宽吸收带。 从C-CDs@K0.3Bi0.7F2.4的 荧 光 光 谱 图 可 见 :C-CDs@K0.3Bi0.7F2.4在450 nm 蓝 光 激 发 下,在555 nm 处呈现出宽带黄色发射峰;此外,在激光波长为400—450 nm 之间时,随着激发波长的增加,其发射强度逐渐增大且发射波长的峰位基本保持不变;在激发波长为450 nm 时,发射强度最大;当激发波长为450—490 nm 之间时,发射强度逐渐降低且发射波长的峰位发生轻微红移,这是具有激发依赖性CDs 普 遍 存 在 的 现 象。 与C-CDs 相 比,CCDs@K0.3Bi0.7F2.4纳米复合材料的发射峰发生了红移,这是因为CDs 与Bi3+离子之间形成共价键导致的。

图2 C-CDs 和C-CDs@K0.3Bi0.7F2.4的光学性能图Figure 2 Optical properties of C-CDs and C-CDs@K0.3Bi0.7F2.4
2.2 C⁃CDs 和C⁃CDs@K0.3Bi0.7F2.4纳米复合材料晶体结构和形貌表征
图3 为K0.3Bi0.7F2.4和C-CDs@K0.3Bi0.7F2.4纳 米复合材料的XRD 图谱[22]。从图3 可见,K0.3Bi0.7F2.4和C-CDs@K0.3Bi0.7F2.4的XRD 图谱基本保持一致,都可以很好地与六方相 K0.3Bi0.7F2.4的标准卡JCPDS#84-0534 相 对 应 , K0.3Bi0.7F2.4和C-CDs@K0.3Bi0.7F2.4均 在26.2、30.3、43.4、51.4、53.9、63.1 和69.5 °处 存 在 衍 射 峰,分 别 对 应K0.3Bi0.7F2.4的(111)、(200)、(220)、(311)、(222)、(400)和(331)晶 面 。 与 K0.3Bi0.7F2.4相 比 ,C-CDs@K0.3Bi0.7F2.4纳米复合材料的XRD 谱图没有明显的变化,说明CDs 的引入对C-CDs@K0.3Bi0.7F2.4的晶体结构没有影响。此外,从C-CDs@K0.3Bi0.7F2.4纳米复合材料中没有观察到C-CDs的特征衍射峰,可能是因C-CDs 的含量较少而导致峰较弱,并且与K0.3Bi0.7F2.4基质衍射峰重叠在一起[22]。

图3 K0.3Bi0.7F2.4和C-CDs@K0.3Bi0.7F2.4的XRD 图Figure 3 XRD patterns of K0.3Bi0.7F2.4 andC-CDs@K0.3Bi0.7F2.4
为了进一步研究C-CDs@K0.3Bi0.7F2.4纳米复合材料的微观形貌,首先对合成的C-CDs 进行了形貌结构表征,图4 为C-CDs 纳米颗粒的TEM 图和HRTEM 图。从 图4 可 见:C-CDs 呈 球 形 颗 粒 且 分散均匀,没有发生团聚现象;C-CDs 的晶格间距为0.21 nm,对应于石墨烯的(100)晶面,表明成功合成了结晶性良好的C-CDs。

图4 C-CDs 的TEM 和HRTEM 图Figure 4 TEM and HRTEM of C-CDs
其次,对K0.3Bi0.7F2.4和C-CDs@K0.3Bi0.7F2.4纳米复合材料的形貌进行了表征,图5 为K0.3Bi0.7F2.4和C-CDs@K0.3Bi0.7F2.4的SEM 和HRTEM 图。 从SEM 图可见:K0.3Bi0.7F2.4具有良好的分散性且形貌呈类球形颗粒,粒径大小均匀;而C-CDs@K0.3Bi0.7F2.4纳米复合材料的貌呈类球形并且团聚堆积在一起,粒径大小不均匀,表明C-CDs 可能附着在K0.3Bi0.7F2.4上,导致粒径发生变化。从HRTEM 图可见:K0.3Bi0.7F2.4的(111)晶面晶格间距为0.33 nm,与立方相K0.3Bi0.7F2.4很好地吻合;而C-CDs@K0.3Bi0.7F2.4的(111)晶面的晶格间距为0.33 nm,(100)晶面的晶格间距为0.21 nm。SEM 和TEM 分析证实了CCDs 在K0.3Bi0.7F2.4表面的成功引入。

图5 K0.3Bi0.7F2.4及C-CDs@K0.3Bi0.7F2.4的SEM 图和HRTEM 图Figure 5 SEM image and HRTEM image of K0.3Bi0.7F2.4 and C-CDs@K0.3Bi0.7F2.4
图6 为C-CDs@K0.3Bi0.7F2.4纳米复合材料EDS的元素分布图。从图6 可以看出,K、Bi 和F 元素均匀地分布在整个纳米颗粒上,而C 和O 元素分布不均匀,说明纳米复合材料中存在CDs,这一结果与上述SEM 和TEM 结果一致。

2.3 C⁃CDs@K0.3Bi0.7F2.4纳米复合材料结构分析
为了探究C-CDs@K0.3Bi0.7F2.4纳米复合材料的元素组成和化学结构,对其进行了FT-IR 和XPS 的表征。
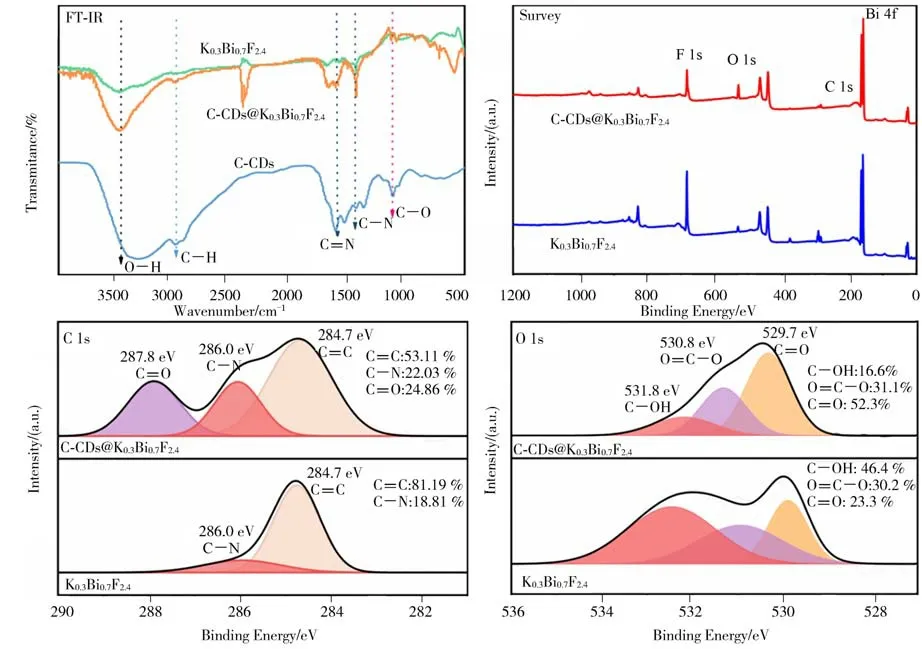
图7 C-CDs、K0.3Bi0.7F2.4和C-CDs@K0.3Bi0.7F2.4的FT-IR、XPS 光谱图Figure 7 The FT-IR spectra and XPS survey spectra of C-CDs,K0.3Bi0.7F2.4 and C-CDs@K0.3Bi0.7F2.4
图7 为C-CDs、K0.3Bi0.7F2.4和C-CDs@K0.3Bi0.7F2.4纳米复合材料的FT-IR 和XPS 光谱图。从图7 中的FT-IR光谱图可见:K0.3Bi0.7F2.4和C-CDs@K0.3Bi0.7F2.4纳米复合材料分别在3150—3550 cm−1及2940、1387 和1063 cm−1峰位存在特征峰,他们分别对应O—H/N—H 的伸缩振动、C—H 不对称伸缩振动、C—N 伸缩振动及C—O 伸 缩振动;此外,与K0.3Bi0.7F2.4相比,C-CDs@K0.3Bi0.7F2.4纳米复合材料特征峰强度明显增强,这一结果再次证实了C-CDs与K0.3Bi0.7F2.4复合在一起。从图7 中K0.3Bi0.7F2.4和C-CDs@K0.3Bi0.7F2.4纳米复合材料的全光谱图可见,谱图中有明显的Bi 4f、F 1s、C 1s、O 1s 的衍射峰,说明二者的主要是由Bi、F、C 和O 元素构成。从图7 中C 1s 的高分辨率XPS 光谱可见,在284.7、286.0 和287.8 eV 处有3 个主峰,分别属于C═C、C—N 和C═O 基团;与K0.3Bi0.7F2.4相比,C-CDs@K0.3Bi0.7F2.4纳米复合材料中C—N 峰面积增加,并出现了新的C═O 基团,表明C-CDs@K0.3Bi0.7F2.4纳米复合材料中存在C-CDs。从图7 中O 1s 高分辨率XPS 光谱可见,在529.7、530.8 和531.8 eV 处有3 个主峰,分别 属 于C ═O、O ═C—O 和C—OH 基 团;在C-CDs@K0.3Bi0.7F2.4纳米复合材料中,由于C-CDs表面存在羧基官能团,这促进了C═O 基团比例的增加,表明C-CDs 在K0.3Bi0.7F2.4表面成功修饰。
2.4 发光机理分析
从紫外吸收光谱可知C-CDs 在300—400 nm的宽吸收带归属于C ═O 键,并且吸收带位置与C-CDs 溶 液 位 于420 nm 的 宽 激 发 峰(350—450 nm)部分重合。综合上述结果与分析,C-CDs 的发光主要来自于表面态发光[23]。此外,K0.3Bi0.7F2.4颜色为白色且无荧光,而所制备的C-CDs@K0.3Bi0.7F2.4的颜色为米黄色且发射出555 nm 的黄色荧光。这是因为C-CDs 表面富含羧基官能团导致C-CDs 表面带有负电荷[24-25],可以通过静电相互作用吸引基质中Bi3+离子。因此,C-CDs 的表面必然发生变化,从而影响C-CDs 表面电子结构而导致能隙减小,吸收带红移。
2.5 C⁃CDs@K0.3Bi0.7F2.4 纳米复合材料的热稳定性研究
荧光粉在高温下的发光稳定性,通常被认为是评价其应用潜力的重要标准。基于上述分析,进一步测量了在λex=450 nm 激发下C-CDs@K0.3Bi0.7F2.4纳米复合材料的热稳定性,结果如图8 所示。从图8中的随温度变化的发射光谱可见,当温度从25 ℃升高到125 ℃时,C-CDs@K0.3Bi0.7F2.4纳米复合材料的发射峰曲线形状和峰位基本保持不变,表明随着温度的升高C-CDs@K0.3Bi0.7F2.4纳米复合材料的发射中心没有受到破坏,由于无辐射跃迁增加,导致发射强度降低。从图8 中不同温度下归一化荧光强度图谱可见,C-CDs@K0.3Bi0.7F2.4纳米复合材料的荧光强度在125 ℃下还能保持初始强度的55%。相比于C-CDs 溶 液 在 高 温 下 荧 光 猝 灭 而 言 ,C-CDs@K0.3Bi0.7F2.4纳米复合材料具有较好的热稳定性。

图8 不同温度下C-CDs@K0.3Bi0.7F2.4纳米复合材料的热稳定性Figure 8 Thermal stability of C-CDs@K0.3Bi0.7F2.4 nanocomposites at different temperatures
3 结论
采用一种简单快速的共沉淀方法,制备出了基于C-CDs的长波长固态发光材料,C-CDs@K0.3Bi0.7F2.4为发射波长位于555 nm 的黄色荧光纳米复合材料,其可有效抑制C-CDs 的聚集诱导荧光猝灭。C-CDs@K0.3Bi0.7F2.4的发射波长与C-CDs 溶液波长相比发生了红移,红移的原因是引入Bi3+离子后C-CDs 的表面状态可产生新的黄色发射。基于化学成分和结构分析,证明了C-CDs 成功地修饰在K0.3Bi0.7F2.4纳米粒子上。此外,C-CDs@K0.3Bi0.7F2.4纳米复合材料具有温度依赖性的黄色发射行为,表明纳米复合材料具有较好的热稳定性。
