基于图神经网络的药物相互预测方法
2022-08-31马怡青蔡美玲陈明成明俊
马怡青 蔡美玲 陈明 成明俊


摘要:药物-药物相互作用(Drug-drug interactions, DDIs)指病人在一定时间内服用两种及以上药物后药物产生的复合效应,可表现为药性增强或减弱。本文提出一种基于图神经网络模型的预测方法,在已有药物间相互作用基础上,结合药物化学结构特征等属性,分进行药物间相互作用预测实验。
关键词:药物相互预测;图神经网络;符号网络;深度学习
中图分类号:TP391 文献标识码:A
文章编号:1009-3044(2022)18-0061-03
开放科学(资源服务)标识码(OSID):
1 引言
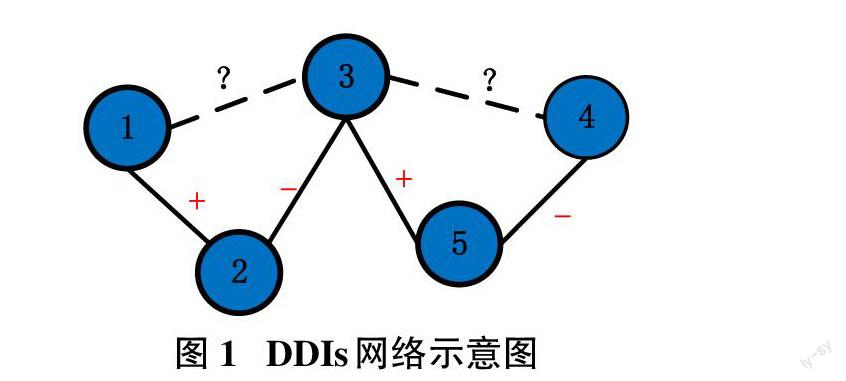
药物-药物相互作用(Drug-drug interactions, DDIs)指的是两种及以上药物混合使用时药物产生的副作用效果[1],具体可表现为药效增强或药效减弱作用。这种现象在现实用药中表现为药性、毒性等增强或减弱。如果将药物间的增强关系视为是正向作用,减弱关系视为是负向作用,那么药物间相互作用关系可以使用一张图的邻接表进行表示。
假定使用图[G(D,E)]来表示一个DDIs网络,其中[D=d1,d2,...,dm]集合, 表示大小为[m]的药物结点[d]集合;[E]表示药物结间关系,如图1所示。图中,结点1和结点2间存在已证明的增强关系;结点2和结点3间存在已证明的减弱关系,而结点1和结点3间的关系是未知。通常,图[G(D,E)]可以用一个[m×m]的邻接对称矩阵[Am×m=aij]表示,如式(1)所示,1表示藥物结点间存在增强关系,-1则为减弱关系,0则为未知。预测实验是根据此邻接矩阵中的非0元素信息,对0值元素预测。
[A=0100010-1000-10010000-1001-10] (1)
2 相关工作
对于药物间相互作用预测,生物实验虽然准确,但成本高、耗时久。计算方法相反,所以在工业界和学术界取得很大关注。多数现有的基于深度学习的预测方法,主要为传统的二值预测,即预测某一药物对间产生DDIs的可能性。Liu等人利用随机森林预测模型结合多特征进行药物相互作用预测[2];Liu等人基于胶囊网络的动态路由机制对文本中的药物相互作用关系进行抽取[3];Feng等人结合图卷积网络GCN和深度神经网络DNN进行药物相互预测[4]。考虑DDIs更具体的增强或减弱作用,必须综合考虑DDIs网络结构。本文通过使用流行的图神经网络模型—SGCN模型,在式(1)定义的符号网络上进行药物间相互作用预测。
3 基于结点嵌入的DDIs预测框架
结点嵌入的目标是对结点进行编码,生成一种结点的特征向量表示,这种向量表示不仅应体现结点的自身属性,同时应能反应图中结点间邻居关系。考虑到已知的药物间相互作用关系和药物自身的特征表示,本文分两步骤来解决DDIs预测问题,如图2所示:
Step1特征提取:针对给定的邻接对称矩阵[A]和对应结点的特征矩阵[T],通过图神经网络模型,针对所有药物结点得到一种低维向量表示[Z],[F1=A,T→Z]。
Step2相互作用预测:对任何一对药物结点对[di]和[dj],将它们的低维向量表示[Zi]和[Zj][Zi,Zj∈Z]进行拼接作为药物节点对的特征表示,使用线性分类器对[di]和[dj]的相互作用关系进行类别预测,[F2:(Zi,Zj)→0,1,-1]。
本文使用线性回归模型对药物间相互关系类型的进行预测。对药物节点对的特征表示,通过线性分类器将它映射到3种情况{0,1,-1},保留正负情况中较大概率一方作为结果。
3.1 基于SGCN的结点嵌入
传统的图卷积神经网络例如GCN[5],只考虑结点间是否有边,即邻接矩阵元素只考虑0或1,不适用于符号预测。
SGCN理论[6]出自平衡理论。平衡理论认为“朋友的朋友是朋友”而“朋友的敌人是敌人”。假设从结点[i]出发,经过[l]条边可以抵达结点[j],若此路径包含偶数条负边,则[j∈Bi(l)],即结点[j]属于结点[i]的[l]层朋友集合;若此路径包含奇数条负边,则[j∈Ui(l)],即结点[j]属于结点[i]的[l]层敌人集合。
假设[h0i]是结点[i]的原始特征,维度大小为[din]。SGCN模型中结点[i]第一层隐藏层输出分别考虑它的直接朋友和直接敌人的特征表示,定义如式(2):
[hB(1)i=σ(WB(1)[j∈N+ih(0)jN+i,h(0)i]),hU(1)i=σ(WU(1)[k∈N-ih(0)kN-i,h(0)i])] (2)
其中[σ]是一个非线性激活函数,在实现中选用ReLu函数。[WB1,WU1∈Rdoutx2din]是模型分别针对结点[i]的直接朋友集合[Bi(1)]和直接敌人集合[Ui(1)]的变换矩阵,[dout]是隐藏层输出向量的长度。
结点[i]的第[l]层表达以迭代方式同时考虑[l-1]层邻结点的朋友集合、敌人集合和自身表达。聚合公式如式(3):
[hB(l)i=σ(WB(l)[j∈N+ihB(l-1)jN+i,k∈N-ihU(l-1)kN-i, hB(l-1)i]), ]
[hU(l)i=σ(WU(l)[j∈N+ihU(l-1)jN+i,k∈N-ihB(l-1)kN-i,hU(l-1)i])] (3)
其中[ WBl,WUl∈Rdoutx3din]。
经过两次聚合,可以获取药物结点[i]的两个隐藏层表示,称为[hB(2)i]和[hU(2)i],这里将这两个隐藏层输出进行拼接作为结点
4 实验
4.1实验数据集
本文使用了来自四篇DDIs预测相关性的论文中提供的不同数据,包括4个数据集,如表1所示。其中, DB1有603个药物节点[1]; DB2有568个药物节点[7]; DB3有1562个药物节点,附有881维的化学结构特征和1642维的药物捆绑蛋白质结构特征[8],都是二值信息; DB4[4]有1934个药物节点。
4.2实验设置
本文选取了通用的评价指标来对本文提出的算法进行性能评估,包括精确度(accuracy)、准确率(precision)、召回率(recall)、[F1]指标和AP指标,同时还计算AUC和AUPR值。
以上指标中,有一部分计算结果是针对模型的预测得分。本文考虑两种角度的预测打分,一类是根据线性分类器得到的预测概率进行计算打分(连续值),一类是将对线性分类器得到的预测概率进行处理,选择正负边中概率较大的一方作为最终预测结果作为分数。
5 实验结果
5.1 SGCN参数调谐
参数设置上,本文使用格点搜索的方法,寻找最优指标对应的参数。降低后的维度分别为:8、16、32、64、128、256。
综合参数调谐结果,本文后续实验主要基于DB3进行,SGCN最优的参数设置:降低后的维度为256时,实验结果相较于其他参数设置,表现出更好的综合指标结果。
5.2 药物化学结构特征,药物蛋白质结构特征等特征比较
实验基于DB3数据集进行参数比较和指标评估。药物的特征的包括:药物的化学结构特征、药物的蛋白质属性特征以及基于SSE方法生成的药物在网络中产生的结构特征[6]。
实验中发现药物化学结构PubChem特征单独使用时,指标结果差于其他两种特征单独使用情况,药物的蛋白质结构DBP特征和药物在网络中的位置信息SSE特征表现出相似的指标结果。指标结果波动情况也是PubChem在低维度时波动最大,SSE会略优于DBP特征。
同时,实验发现使用线性分类器分类结果作为预测分数时计算AUC指标和AP指标的结果会更优。
表2是三种特征单独使用,在最优参数设置下的各指标结果:
其中auc1表示使用预测结果作为预测分数时AUC指标的值,auc2则表示使用线性分类器分类分数作为预测分数时AUC指标的值。以此类推其他指标。
本文还考虑通过拼接这些特征得到更多的药物信息进行DDIs预测实验,这里使用最好预测效果的DBP和SSE进行拼接然后实验,还考虑了将这三者进行拼接进行实验,实验结果如表3所示:
实验指标和单独采用SSE的情况没有太高的优化,侧面反映SSE应该是其中最有效果的特征信息。
表4是采用其他三个数据集得到的实验结果和DB3的汇总,在这些实验中,统一使用了SSE特征作为药物节点的输入特征表示。
6 结束语
本文使用图符号网络模型在公开药物数据集上进行药物相互关系预测实验:通过对于药物结点间正向边和负向边的不同处理聚合药物图中药物邻接点信息,获得药物的嵌入特征表达;通过拼接两个药物结点的特征表达的方法使用线性分类器进行分类预测。在已有公开数据集上的实验结果表明,本文所述方法是有效的,亦是对现有方法的有益补充。
参考文献:
[1] Shi J Y,Huang H,Li J X,et al.TMFUF:a triple matrix factorization-based unified framework for predicting comprehensive drug-drug interactions of new drugs[J].BMC Bioinformatics,2018,19(Suppl 14):411.
[2] 刘光徽,胡俊,於东军.基于多视角特征组合与随机森林的G蛋白 偶联受体与药物相互作用预测[J].南京理工大学学报(自然科学版),2016,40(1):1-9.
[3] 刘宁宁,琚生根,熊熙,等.基于胶囊网络的药物相互作用关系抽取方法[J].中文信息学报,2020,34(1):80-86,96.
[4] Feng Y H,Zhang S W,Shi J Y.DPDDI:a deep predictor for drug-drug interactions[J].BMC Bioinformatics,2020,21(1):419.
[5] Kipf T N, Welling M. Semi-supervised classification with graph convolutional networks[J]. arXiv preprint arXiv:1609.02907, 2016.
[6] Derr T,Ma Y,Tang J L.Signed graph convolutional networks[C]//2018 IEEE International Conference on Data Mining.November 17-20,2018,Singapore.IEEE,2018:929-934.
[7] Yu H,Mao K T,Shi J Y,et al.Predicting and understanding comprehensive drug-drug interactions via semi-nonnegative matrix factorization[J].BMC Systems Biology,2018,12(Suppl 1):14.
[8] Shi J Y,Mao K T,Yu H,et al.Detecting drug communities and predicting comprehensive drug-drug interactions via balance regularized semi-nonnegative matrix factorization[J].Journal of Cheminformatics,2019,11(1):28.
【通聯编辑:唐一东】
