一个Menkes病家系的临床特征及基因变异分析
2022-08-17王越王怀立禚志红
王越,王怀立,禚志红
(郑州大学第一附属医院 儿科,河南 郑州 450052)
Menkes病(Menkes disease,MD,OMIM:309400)是由于ATP7A基因(0MIM:300011)变异导致铜代谢紊乱所致的遗传性神经系统退行性病,呈X-连锁隐性遗传。该病较为罕见,国外不同国家不同时间段内报道的发病率不一,从1/354 507到1/40 000,日本报道大约为1/357 000活产婴儿[1]。目前国内多为少数家系或者个例报道[2],2017年北京大学第一医院总结了24例男性MD患儿,是目前最大的中国MD患儿数据样本[3]。现本研究对1个MD家系的临床特点及基因变异特点报告如下,并结合近年来国内外的研究进展,探讨该病特点。
1 对象与方法
1.1 研究对象患儿,男,出生日期2020年1月9日,就诊时月龄为3个月,因“发热、抽搐发作10 d”就诊。2个月18天时首次出现抽搐发作,表现为头偏向右侧,双眼向右凝视,四肢强直,双手握拳伴抖动,面色发绀、口吐白沫,持续3~5 min缓解,伴发热,热峰38.5 ℃,抽搐发作每日2~3次并逐渐增加至每日3~5次。患儿为第1胎,第1产,胎龄37周,顺产娩出,出生体质量2 700 g,出生时因窒息在NICU抢救。母孕期无异常。生后人工喂养,喂养困难。父母体健,非近亲结婚,否认家族遗传病史。查体:发育落后,不能竖头、追物,逗笑无反应,哭无力,面颊饱满(图1),头发稀疏、色黄、卷曲且出生即有。肤色偏白,皮肤干燥、松弛、弹性差。四肢肌张力低,腱反射活跃,巴氏征阴性,肌肉松弛、无力。血常规、血氨、血乳酸正常,脑脊液常规、生化、病毒抗体、培养结果均正常。血清铜蓝蛋白4.13 mg·dL-1(参考范围15~45 mg·dL-1),血清铜7.4 μmol·L-1(参考范围8.51~25.63 μmol·L-1),均降低。头颅MRI:左侧额颞部硬膜下积液(图2)。视频脑电图:醒睡各期左右半球均可见大量尖波、尖慢波、棘慢波发放,左右同步或不同步发放。
临床拟诊为:MD,应用左乙拉西坦口服液癫痫发作控制不佳,加用丙戊酸钠口服液后癫痫发作次数逐渐减少至无发作。现口服左乙拉西坦与丙戊酸口服液维持,患儿10个月龄时电话随访,无癫痫发作,智力、运动发育出现倒退,反复出现肺部感染。
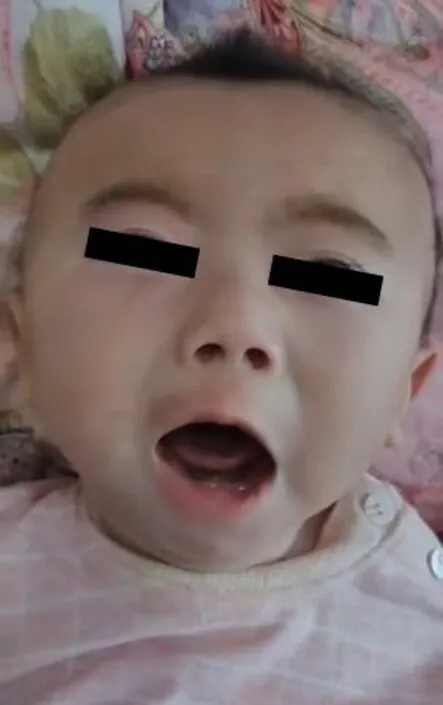
图1 患儿面容及头发

图2 患儿头颅MRI平扫提示额颞部硬膜下积液
1.2 方法
1.2.1DNA提取 本研究经医院医学伦理委员会批准(2021-KY-0149-002),经患儿父母同意,抽取患儿及其父母静脉抗凝血,使用全血DNA核酸提取试剂盒提取EDTA抗凝外周血基因组DNA(gDNA),-20 ℃保存。
1.2.2全外显子测序 将gDNA打断,补平DNA末端,加“A”尾和接头,构建测序文库,使用探针对gDNA进行基因组全外显子片段捕获,进行双端高通量测序。
1.2.3生物信息学分析 利用Bcl2fastq(v2.0.1)工具将测序原始数据文件转化为fastq文件,过滤接头、低质量序列,统计测序深度、覆盖度等信息。使用工具将数据与hg19基因组比对。基于OMIM、ClinVar、HGMD和gnomAD等数据库及人群大规模测序数据库,对变异进行筛选,并利用生信预测软件进行变异有害性预测,生成遗传分析数据。
1.2.4遗传分析 将注释后数据过滤掉人群多态性位点,查阅OMIM、Clinvar、HGMD数据库,结合美国医学遗传学与基因组学会遗传变异分类标准与指南[4-5],综合判断变异的致病性。
1.2.5一代测序验证 根据二代测序数据设计合成测序所需引物,进行PCR扩增并Sanger测序,测序结果与全外显子测序后的结果进行比对。ATP7A基因上游引物5’-ATAGATGATAGCCCCACAGGC-3’,下游引物5’-CAGCATAGGGGGTGTGTCAAA-3’。
2 结果
通过全外显子测序及分析,发现患儿X染色体ATP7A基因(NM_000052)存在半合子变异,为c.2284_2285dupTT(p.Leu762Phefs*2),位于第10号外显子上,为移码变异,预计会使变异型的ATP7A蛋白在第762位氨基酸由亮氨酸(Leu)变为苯丙氨酸(Phe),造成该位点之后的蛋白质三联体密码子阅读框发生改变(图3)。Sanger测序验证家系共分离,结果显示父亲未携带上述变异,而母亲携带(图4),即患儿变异遗传自母亲。ATP7A基因的c.2284_2285dupTT(p.Leu762Phefs*2)变异在dbSNP、gnomAD、ExAC、千人基因组G1000、ESP6500人群频率数据库中均未收录,表明该变异频率在全球人群极为罕见。数据库HGMD、Clinvar均未见报道,通过PubMed文献检索亦未见该变异的相关报道。先证者存在ATP7A基因半合子变异,符合X染色体隐性遗传疾病发病机制,结合先证者与家系成员符合家系共分离,该患儿被诊断为MD,最终确定ATP7A基因为先证者的致病基因。

图3 患儿二代测序可视化图谱 ATP7A基因c.2284_2285dupTT

图4 Sanger测序图谱
3 讨论
MD又称卷发综合征,是一种罕见的由ATP7A基因变异引起的X连锁隐性遗传病。该病最早由Menkes等[6]在1962年报道。ATP7A基因编码的蛋白称为ATP7A酶,是一种P型跨膜铜离子转运蛋白,该蛋白主要在肠黏膜上皮细胞中表达,其作用是将肠黏膜上皮细胞内的铜排至细胞外液供机体利用。ATP7A基因变异导致ATP7A酶功能缺陷,影响肠黏膜铜的吸收,导致机体铜缺乏,降低铜相关酶的活性,从而引起多系统功能障碍[7]。
根据临床表现不同,MD可分为经典型、轻型、枕角综合征,其中经典型患儿占90%~95%。绝大多数为男性患者。经典型MD在婴儿期发病,主要临床表现为特征性卷发、进行性加重的神经系统退行性变性、结缔组织异常等。新生儿期因临床表现不特异容易被忽视。国外报道新生儿期的临床特征包括黄疸时间延长、体温过低、肌张力减退、低血糖和出生时喂养困难等。国内有研究表明中国MD患儿中早产和窒息常见。本例患儿为男性,出生时有窒息史,且有喂养困难,与上述报道一致。特征性的皮肤毛发表现为肤白、头发稀疏、颜色浅、容易断、发量少,主要分布在头顶部[8]。本例患儿出生时即出现头发稀疏、色浅、卷曲。结缔组织异常表现为皮肤干燥、松弛、腹股沟疝、脐疝、膀胱扭转、肌张力低下、关节过度活动或脱位,血管走形迂曲,累及全身各处血管,以颅内血管为著[9],颈内静脉扩张及肱动脉动脉瘤均有报告。骨骼异常表现为骨畸形、骨质减少和长骨骨折。干骺端骨折、肋骨骨折和颅骨骨折常被误诊为虐待儿童[10]。本研究患儿皮肤干燥、松弛、弹性差,四肢肌张力低下,肌肉松弛无力,与文献报道一致。
神经系统受累表现为癫痫发作、神经发育迟滞、倒退。一般在2~3个月左右开始出现发育迟滞、癫痫发作,感染或发热会触发癫痫发作。癫痫发作可分为3期[11],表现形式多样,较难治,多种抗癫痫发作药物联合应用控制不佳,脑电图呈中至重度异常。本研究患儿癫痫起病2个月18天,起病前出现发热,抽搐发作形式、脑电图特点均与上述报道相符。随访发现患儿神经系统发育迟滞严重,并出现倒退,提示颅内病变较重,但是该患儿口服2种抗癫痫发作药物后未再有抽搐发作,与其他临床表现不平行。国内也有类似报道[8],考虑可能与严重脑萎缩及硬膜下积液有关。但是本研究患儿后期并未再次复查头颅MRI平扫影像学检查,尚不能明确颅内是否有严重脑萎缩病变。
血清学检查表现为血清铜蓝蛋白、铜减低。本研究患儿血清铜蓝蛋白和铜均减低,符合MD特征。但是上述指标在6个月以下的婴儿中通常也偏低,也导致MD新生儿期早期诊断面临困难。国外报道分析血浆中儿茶酚胺可以较准确地筛查新生男婴中MD患儿[12]。影像学检查在MD诊断中有重要作用,典型影像学改变为:颅内血管迂曲、脑白质异常、脑萎缩、硬膜下积液等,其中颅内血管迂曲是MD最突出的表现之一,具有诊断意义[13]。该患儿早期行头颅MRI检查发现单侧硬膜下积液,符合上述改变,但是未进行头颅MRA检测以及后期随访未动态观察头颅MRI改变,缺乏血管改变资料。
确诊MD依靠基因检测。ATP7A基因变异导致蛋白功能缺失,是引起MD的根本原因。ATP7A与ATP7B蛋白是细胞跨膜运输铜的特异性ATP酶,与铜的分泌途径有关[14]。铜本身是机体代谢途径中关键酶所必须的元素,其代谢异常时容易对细胞产生毒副作用,影响儿茶酚胺的生物合成和结缔组织的成熟,从而导致严重的疾病[15-16]。ATP7B主要在肝表达,而ATP7A在除肝外的几乎所有器官表达。 因此,ATP7B变异导致的Wilson疾病主要影响肝,而MD则是全身性疾病[17]。
ATP7A基因位于染色体Xq21.1,包括23个外显子,分布在大约140 kb的基因组DNA上。目前ATP7A被LOVD开放数据库收录了452个公开报道的基因变异(https://databases.lovd.nl/shared/genes/ATP7A),以错义变异为主。Kaler等[18]对gnomAD人群数据库的突变类型统计发现约43%为错义突变,剪切突变、移码突变、无义突变等均在人群中频率小于0.2%,提示稀有变异难以在人群中存活,而致病性较强。本研究通过全外显子测序及分析发现患儿ATP7A基因变异位于第10号外显子上,为c.2284_2285dupTT,属于移码变异,预计会导致蛋白质从第762位氨基酸发生翻译紊乱,可能形成截短蛋白,从而导致ATP7A蛋白功能缺陷,导致肠黏膜铜吸收障碍,使体内广泛性铜缺乏,从而引起多系统功能障碍。研究表明,75%患者的母亲是携带者,其余25%是新生突变[18]。本研究患儿存在半合子变异,通过对患儿父母进行一代Sanger测序验证,父亲未携带上述变异,而母亲携带,即患儿变异遗传自母亲。目前该突变类型国内外尚未见报道。
虽然大多数经典型在3岁内死亡,但是部分持续治疗的患儿可以存活到十几岁[19]。因此,MD早期诊断、早期治疗特别关键。目前尚无根治性手段,口服铜治疗是无效的,皮下或静脉途径给予组胺铜治疗被证明安全有效。国外研究显示,如果在婴儿3周以内就开始治疗,某些患者的神经退行性变可以得到有效的减轻。组氨铜治疗可以有效逆转皮肤和头发的变化,但是治疗结果取决于治疗的开始时间和残存的ATP7A功能。只有在新生儿期开始治疗,组氨酸铜治疗才能有效地提高生存率和减轻疾病的神经负担[12]。国内尚无组氨铜药物,尚缺乏治疗资料。
MD是一种ATP7A基因变异导致的X连锁隐性遗传病。经典型MD患者在婴儿期发病,主要临床表现为特征性卷发、神经系统退行性变性、结缔组织异常,多在3岁内死亡。本研究利用全外显子测序发现ATP7A基因第10外显子核苷酸第2 284~2 285位碱基存在c.2284_2285dupTT移码突变,为致病变异,且国内外未见报道,丰富了ATP7A基因的变异谱,有助于深入了解该基因的结构与功能。
