高效液相色谱法同时测定牛奶中阿苯达唑、氟苯达唑、噻苯达唑及4种代谢物残留量
2022-08-12赵雪宁雷浩贺习文高勤叶李易轩
赵雪宁,雷浩,贺习文,高勤叶,李易轩
(陕西秦云农产品检验检测股份有限公司,渭南 714000)
阿苯达唑、氟苯达唑和噻苯达唑同属于苯并咪唑类药物。苯并咪唑类药物是含有2个氮原子的芳香杂环,以苯并咪唑环构筑的药物分子,具有广泛的生物活性,可作为组胺受体拮抗剂、抗高血压药、广谱驱虫类药、抗真菌药物、抗病毒类药物等。阿苯达唑、氟苯达唑和噻苯达唑均作为广谱驱肠虫类药物被广泛使用[1,2]。
苯并咪唑类药物虽然具有高效低毒的优点,但在实验动物和靶动物中有致畸和致突变的作用,且在动物体内转化的代谢物具有毒理性,所以在动物源性食品中是重点监控的对象[3,4]。目前有关苯并咪唑类药物检测方法的报道多见于动物组织[5]、动物血清和饲料等基质样品,测定乳制品的报道相对较少。汤娟等[6]利用高效液相色谱-串联质谱法测定了奶粉中3种苯并咪唑类药物残留,吴宇鹏等[7]利用UPLC-MS/MS法测定了牛奶中14种苯并咪唑类药物及部分代谢物残留,吴鹏等[8]研制了能测定噻苯咪唑的ELISA检测试剂盒。这些报道中大都使用液相色谱-串联质谱仪,检测成本较高,而ELISA虽然操作快捷简单,但通常仅能筛查一种化合物,具有较大的局限性。
目前,常用的苯并咪唑检测方法有高效液相色谱法[9~11]、ELISA法[8]、液相色谱-串联质谱法[12~17]。本研究采用经济实用,同时能够准确定性的高效液相色谱法,通过调整仪器条件和样品处理过程,建立了一种能同时测定牛奶中3种苯并咪唑类药物和4种代谢物的完整方法,该方法前处理快捷,检出限低,回收率高,稳定性良好,具有广泛使用的价值。
1 材料与方法
1.1 仪器、试剂及材料
Ultimate 3000高效液相色谱仪(Thermo Fisher Scientific公司,美国);H1850型离心机(湖南湘仪实验室仪器开发有限公司);MS205DU型十万分之一天平(Mettler Toledo公司);CP224C型万分之一天平(奥豪斯仪器(常州)有限公司);MX-S型涡旋混合仪(大龙兴创仪器(北京)股份有限公司);KQ-100DB型数控超声波提取仪(昆山市超声仪器有限公司);MTN-4800A型氮吹仪(天津奥特赛恩斯仪器有限公司)。
阿苯达唑(坛墨质检,纯度97.2%);阿苯达唑砜、阿苯达唑亚砜(阿尔塔科技有限公司,纯度98.0%);氟苯达唑、2-氨基氟苯达唑,噻苯达唑、5-羟基噻苯达唑(Dr.Ehrenstorfer,纯度99.3%);乙腈、甲醇(色谱纯,美国Sigma公司);超纯水(杭州娃哈哈集团有限公司);盐酸(优级纯,西陇科学股份有限公司);氨水(25%优级纯)、冰乙酸(分析纯)(天津市富宇精细化工有限公司);2%乙酸溶液;10%盐酸溶液。
C18固相萃取柱、HLB固相萃取柱、MCX固相萃取柱,均为200mg/3mL(月旭科技有限公司)。
1.2 试验方法
1.2.1 色谱条件
色谱柱:Hypersil GOLD C18色谱柱(250×4.6mm,5μm);柱温:30℃;流动相:A相为甲醇,B相为2%乙酸溶液,0~10min 60%B,10~12min 60%B~30%B,12~17min 30%B,17~20min 30%B~60%B;流速:1.0mL/min;检测波长:292nm;进样量:10μL。
1.2.2 样品前处理过程
提取:准确称取5.00g牛奶试样,置于50mL具塞离心管中,加入10mL乙腈,涡旋混合1min,超声提取5min,8 000r/min离心3min,准确移取上清液5.00mL于10mL离心管中,40℃下氮气吹干,加入3mL 10%盐酸溶液复溶,待净化。
净化:取C18固相萃取小柱,依次用3mL甲醇、3mL水活化,将上述净化液全部转移至固相萃取小柱中,控制流速不超过1mL/min,待样液全部通过固相萃取小柱后,用3mL水淋洗,抽干,用5mL甲醇洗脱,收集洗脱液,在40℃下氮气吹干,加入1mL甲醇复溶,过0.45μm滤膜后供高效液相色谱仪测定。
1.2.3 标准溶液的配制
标准贮备液的配制:准确称取阿苯达唑、阿苯达唑砜、阿苯达唑亚砜、2-氨基氟苯达唑、噻苯达唑、5-羟基噻苯达唑标准物质各10mg,分别置于10mL棕色容量瓶中,用甲醇溶解并定容至刻度,摇匀,即得浓度为1mg/mL的6种化合物的标准贮备液;准确称取氟苯达唑标准物质10mg于10mL棕色容量瓶中,用10%盐酸溶液溶解并定容至刻度,摇匀,即得浓度为1mg/mL的氟苯达唑标准贮备液。
标准混合中间液的配制:准确移取7种标准贮备液各1mL于10mL棕色容量瓶中,用甲醇定容至刻度,摇匀,配制成浓度为100μg/mL的混合中间液。
2 结果与讨论
2.1 流动相的确定
试验过程中发现阿苯达唑和氟苯达唑保留时间较长,其余5种化合物保留时间较短,因此采用水相加有机相梯度洗脱的方式。
在水相的选择上,考察了20mmol/L乙酸铵溶液、2%乙酸溶液和0.1%磷酸溶液,其中乙酸铵溶液的洗脱时间最长,磷酸溶液的峰型尖锐,但2-氨基氟苯达唑和阿苯达唑砜难以分离,综合考察下,乙酸溶液的洗脱时间和分离效果最为理想。
在有机相的选择上,结合所使用的C18色谱柱,优先考虑甲醇和乙腈作为有机相。使用乙腈作为有机相时,5-羟基噻苯达唑保留时间过短,与溶剂倒峰相连,无法进行积分计算,同时在梯度洗脱时由于有机相比例变化,基线浮动明显;使用甲醇作为有机相时,5-羟基噻苯达唑保留时间明显后延,同时梯度洗脱时基线没有明显浮动,因此流动相选择2%乙酸溶液和甲醇梯度洗脱,7种化合物的标准溶液谱图和加标样品图谱见图1。
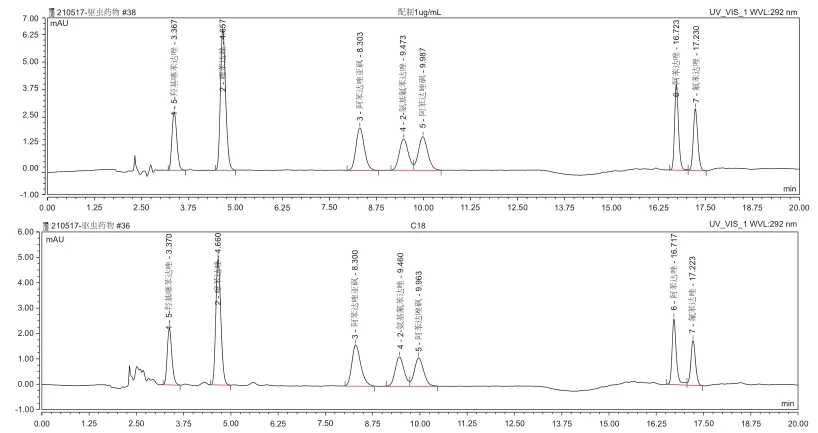
图1 7种化合物的标准溶液谱图和加标样品图谱
2.2 检测波长的确定
使用二极管阵列检测器对7种化合物进行全波长扫描,结果显示最大吸收波长集中在290~310nm之间,使用292nm时7种化合物峰高差距较小。
2.3 提取溶剂的确定
考虑阿苯达唑、氟苯达唑在丙酮和三氯甲烷等有机溶剂中不溶或微溶,因此提取溶剂考虑甲醇和乙腈两种有机试剂。经过试验验证,两种溶剂对除氟苯达唑外的6种化合物均有良好的提取效果。氟苯达唑在甲醇中溶解性差,回收效果低于乙腈。除此之外,乙腈能够沉淀牛奶样品中的蛋白,离心后相比甲醇提取更加澄清,有助于固相萃取柱的净化。
2.4 固相萃取柱的选择
试验考察了HLB、MCX和C18三种不同填料的固相萃取柱,3种固相萃取柱对7种化合物的回收效果见图2。

图2 不同填料固相萃取柱的回收效果
从图2可以看出,HLB填料和C18填料的固相萃取柱回收效果明显优于MCX填料固相萃取柱,C18固相萃取柱对氟苯达唑的回收效果优于HLB固相萃取柱,综合考虑,采用C18固相萃取柱作为净化材料。
2.5 方法学考察
2.5.1 曲线方程和线性范围
使用1.2.3中的混合标准中间液配制成一系列的混合标准工作液,以最优的色谱条件将系列混合标准工作液上机测试,以化合物浓度作为横坐标,以化合物峰面积作为纵坐标绘制成标准工作曲线,所得曲线方程、相关系数和线性范围见表1。

表1 目标化合物的曲线方程、相关系数和线性范围
从表1可以看出,7种化合物在0.05~100μg/mL范围内呈现良好的线性关系,相关系数均≥0.9998,满足试验要求。
2.5.2 检出限与定量限
试验以3倍信噪比作为检出限(LOD),以10倍信噪比作为定量限(LOQ),通过向空白样品中添加目标物的方式来确定检出限和定量限值。所测得的结果见表2。

表2 目标化合物的检出限、定量限、回收率和精密度
从表2可以看出,噻苯达唑检出限为0.025mg/kg,定量限为0.10mg/kg,5-羟基噻苯达唑等6种化合物检出限为0.05mg/kg,定量限为0.20mg/kg。
2.5.3 回收率试验
使用空白样品分别进行了检出限、定量限以及10倍检出限的添加回收试验,试验所得结果见表2。从表2可以看出,7种化合物的回收率在68.9%~94.5%之间,回收效果良好。
2.5.4 稳定性试验
在回收率试验的基础上,将每个组别再同时做6平行的添加试验,以测定的试验结果计算7种化合物的日内变异系数,并将样品保存于4℃阴暗处,连续测定3d,记录试验结果,计算7种化合物的日内和日间变异系数,所得结果见表2。从表2可以看出,7种化合物的日内变异系数在0.7%~3.4%之间,日间变异系数在1.5%~4.8%之间,证明该方法稳定性良好。
3 结论
本研究基于高效液相色谱法和固相萃取技术,建立了一种能同时测定牛奶中阿苯达唑、噻苯达唑、氟苯达唑和4种代谢物残留量的定量分析方法。经过试验论证,方法具有良好的回收率和稳定性,检出限和定量限都很低,且实验成本低廉,操作简单,为牛奶中广谱驱肠虫类药物的测定提供了技术和数据支撑。
