原位构建富氟SEI的凝胶电解质用于金属锂二次电池
2022-08-10李文涛林慧娟钟海
李文涛,林慧娟,钟海
(暨南大学新能源技术研究院,广东 广州 510632)
引 言
随着智能电子产品、新能源汽车、高科技设备等功能日益强大,市场迫切地需要更高能量密度的二次电池以满足其长时间续航的要求。金属锂具有低的标准电极电位(-3.04 V)、高的质量比容量(3860 mAh·g-1)以及低的质量密度(0.534 g·cm-3)等优点,在高能量密度电池中极具应用潜力。将其与常规正极材料配对组成金属锂二次电池(lithium metal rechargeable batteries,LMBs)后,电池的能量密度会有大幅度的提升[1-3]。然而,金属锂的高反应活性会导致其与电解液接触时自发形成SEI,而SEI的稳定性又与电池性能密切相关。同时,金属锂在充放电循环过程中的不均匀溶解沉积会造成持续性的锂枝晶生长,进而导致电池出现安全性问题,严重制约着高能量密度LMBs的商业应用[4-6]。
当前,解决上述问题的方法之一就是采用凝胶聚合物电解质(gel polymer electrolytes,GPEs)来提升LMBs 的循环稳定性和安全性能[7-9]。早期采用非原位聚合方式(如倒相法[10]、Bellcore 法[11]、浇铸法[12]、流延法[13]等)来制备GPEs,所组装电池的电解质/电极界面相容性较差,存在较大界面阻抗,严重影响电池的整体性能[14-15]。因此,研究人员着力于开发原位聚合凝胶电解质的技术,如将前体溶液在引发剂(引发条件)的作用下原位引发单体分子聚合,从而获得GPEs[16]。然而,聚合过程中未反应完的引发剂会影响其与电池各组分之间的电化学稳定性,如偶氮二异丁腈(AIBN)上的富电子—CN 基团与金属锂负极兼容性差且易形成不良的SEI[17],过氧化二苯甲酰胺(BPO)等会热分解产生气体[18]从而影响电池的循环性能。基于此,研究人员进一步研发出不额外加入引发剂制备GPEs 的方法,如使用电化学原位聚合的方式制备聚二氧环戊烷[19],以及使用电解液中的LiPF6引发开环聚合获得聚二氧环戊烷[20-22]。而与1,3-二氧环戊烷结构较为类似的四氢呋喃作为凝胶聚合物网络的研究相对较少[23]。此外,在锂负极表面构建富含LiF 的SEI 来提升LMBs 的长周期稳定循环性能[24-26],一直以来都是金属锂负极保护的研究热点之一。这些研究大多通过引入外来的氟源对锂负极表面进行相应的处理,忽略了商业电解液中LiPF6电解质本身就可以作为生成SEI 的氟源。
本文利用商业电解液中的LiPF6作为导电电解质、聚合引发剂以及氟源,优化参数研制一种原位聚合的凝胶电解质(GPE),采用多种测试方法和手段探究了凝胶电解质的理化性能;并将最优比例的凝胶电解质组装成LMBs,分析其电化学性能,提出改善金属锂二次电池性能的机制原理。为构建无锂枝晶生长、循环稳定的金属锂二次电池提供相应的参考和指导。
1 实验材料和方法
1.1 实验药品
四氢呋喃(超干溶剂,H2O<50×10-6)购自河北百灵威超精细材料有限公司。335C 电解液(国泰华荣,电池级)购自张家港市国泰华荣化工新材料有限公司。磷酸亚铁锂(LiFePO4,电池级)购自深圳贝特瑞新能源有限公司。导电炭黑(SP,电池级)购自苏州晟尔诺科技有限公司。聚偏氟乙烯(PVDF,电池级)购自法阿科玛化学有限公司。金属锂(直径15.6 mm,厚度为45 μm)购自天津中能锂业。
1.2 凝胶聚合物电解质的制备
按质量比1∶2、1∶3、1∶4 称量THF 溶剂与335C电解液,加到玻璃瓶中密封并静置。所使用的335C电解液为碳酸乙烯酯(EC)、碳酸甲乙酯(EMC)和碳酸二甲酯(DMC)的混合物溶剂(1∶1∶1,体积比)中含有1 mol·L-1的LiPF6的电解液。以上所有操作均在氩气填充的手套箱中进行(H2O 和O2<1×10-6)。纤维素支撑的GPE 是将纤维素片放入静置24 h 后的混合溶液中,使其充分浸润,待再次静置24 h 后取出使用。PTHF 是由1 mol·L-1LiPF6/THF 前体溶液自聚合制备而成。
1.3 电池组装及电化学测试
采用金属锂为负极、LiFePO4为正极,纤维素隔膜为支持膜,注入50 μl 的GPE 前聚体溶液组装CR2032 的纽扣电池,静置12 h 后再进行充放电测试。其中,正极由活性物质LiFePO4(80%(质量))、导电剂Super P(10%(质量))和黏结剂聚偏二氟乙烯(PVDF,10%(质量))组成,正极极片直径为12 mm,活性物质载量约为2 mg·cm-1。充放电使用的仪器为LAND CT2001A 电 池 测 试 系 统(LAND Co. Ltd.,China)。LMBs 的充放电电压范围为2.0 ~4.0 V,充放电电流密度为1 C(1 C = 150 mA·g-1)。交流阻抗谱(EIS)使用CHI660E 在0.1 Hz~1 MHz 的范围内以5 mV 的振幅采集数据。电化学窗口采用电化学线性扫描(LSV)方式获得,测试范围为-1 ~6 V,扫速为0.1 mV·s-1。通过EIS测试两电极阻塞电池获得离子电导率σ,并根据公式σ=d/SR计算。其中,d为电解质厚度;S为电极与电解质的有效接触面积;R为电解质的本征电阻。锂离子迁移数是通过EIS 和计时电流法组装Li|Li对称电池测试,并根据Bruce-Vincent-Evans(BVE)公式计算:tLi+=Iss(ΔV-IoRo)/[Io(ΔV-IssRss)]。其中,Io为初始电流;Iss为稳态电流;ΔV是外加的10 mV 小极化电压;Ro和Rss分别为极化过程前后的初始界面电阻和稳态界面电阻。
1.4 分析表征
采用傅里叶红外光谱(FTIR)仪分析凝胶聚合物。采用日立S-4800 场发射扫描电子显微镜(FESEM)观察电解质和金属锂表面的微观形貌。采用X 射线光电子能谱(XPS)分析金属锂表面的SEI成分。
2 实验结果与讨论
2.1 GPE的理化性质
图1(a)展示了335C∶THF质量比为1∶3前体溶液转变为凝胶聚合物电解质前后形态的变化以及反应过程。在前体溶液中,少量的LiPF6分解为LiF和PF5,后者在室温下会与THF 形成一种氧 离子[(THF)+(PF5)-][27],这种氧 离子会插入THF的五元环使其开环聚合,随着分子链的增长,最终生成聚四氢呋喃(PTHF)[28]。而335C 电解液则被固定在PTHF 的网络结构中,获得一种不具流动性的凝胶态电解质。同时,质量比为1∶2 和1∶4 的前体溶液作为对照实验组与之进行了对比,如附录图A1 所示。结果表明,放置48 h 后质量比为1∶2、1∶3 和1∶4 的前体溶液中,THF 均能够生成聚合物。但在1∶2 前体溶液中335C 电解液的含量过高,导致PTHF 无法将所有的335C 电解液固定在网络结构中,玻璃瓶倒置后出现较多液体渗出的现象。而1∶4前体溶液中存在聚合物固体与溶液分层的现象。因此,本文选取1∶3 的组分进行进一步的研究。图1(b)为纤维素隔膜支撑的GPE 照片,表观上可以看出含有大量的凝胶电解质。图1(c)、(d)为GPE 的微观形貌图,从图中可以看出GPE 表面非常平整。与空白纤维素隔膜相比[图1(e)],凝胶电解质完全填充在纤维素隔膜的孔隙中。
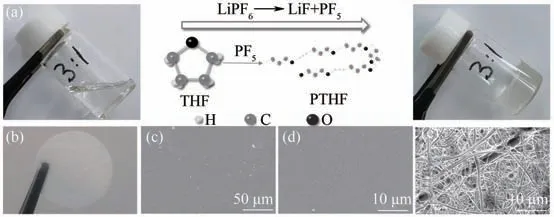
图1 (a)THF开环聚合机理示意图;(b)纤维素隔膜支撑的GPE光学照片;(c),(d)不同放大倍数的GPE的SEM图;(e)纤维素隔膜的SEM图Fig.1 (a)Illustration of polymerization mechanism;(b)Photograph of GPE with cellulose film;(c),(d)SEM images of GPE under different magnifications;(e)SEM image of cellulose film
2.2 GPE电化学性能测试
利用傅里叶红外光谱(FTIR)技术分别对GPE、PTHF 和335C 电解液进行表征[图2(a)]。从图中可以看出,GPE 在1451、1368 cm-1(—CH2弯曲振动)和1105 cm-1(C—O—C 伸缩振动)[29]出现的特征峰与PTHF 的基本一致,表明GPE 中存在PTHF。而位于1751 cm-1和1271 cm-1的吸收峰为335C 电解液中碳酸酯溶剂(—C===== O 与—CH3官能团)的特征峰[30],表明凝胶电解质中含有335C电解液。

图2 (a)GPE,PTHF以及335C电解液的FTIR光谱;(b)GPE与335C电解液的离子电导率-温度变化曲线;(c)Li|GPE|Li对称电池的计时安培曲线(插图为对称电池极化前后的交流阻抗谱);(d)GPE与335C电解液的LSV曲线Fig.2 (a)FTIR spectra of GPE,PTHF and 335C electrolyte;(b)Temperature dependent ionic conductivity of GPE and 335C electrolyte;(c)The chronoamperometry profile of Li|GPE|Li symmetric cell(the inset displays the impedance spectrabefore and after chronoamperometry);(d)Linear sweep curves of GPE and 335C electrolyte
图2(b)展示了GPE 和335C 电解液离子电导率随温度变化的曲线。室温下GPE 的离子电导率为1.33 mS·cm-1,稍低于335C 电解液的1.47 mS·cm-1。随着温度的升高,两者的离子电导率也随之增加,但是GPE 的离子电导率增加幅度明显小于335C 电解液。这可能是由于聚合物网络结构使得335C 电解液无法自由流动,从而降低了温度对GPE 离子电导率的影响。图2(c)为Li|GPE|Li对称电池的计时安培曲线,插图为对称电池极化前后的交流阻抗谱。图A2(a)为Li|GPE|Li 电池的等效电路模型和使用Zview 软件进行等效电路拟合实验的阻抗谱图。由BVE 公式计算出的GPE 锂离子迁移数为0.5。相对于335C 电解液的锂离子迁移数[0.48,图A2(b)]有了稍微的提升。这可能是聚合物网络结构所带来的空间位阻效应对PF6-这种大阴离子迁移过程产生了一定的影响。图2(d)展示了GPE、335C 电解液的LSV测试结果,发现GPE氧化起始电流电位为4.5 V,高于335C 电解液[4.1 V(vsLi/Li+)],能够满足常规正极材料(LiFePO4、LiMn2O4、LiCoO2、NCM 等)[31-32]的正常充放电。图A3 展示了GPE 和335C 电解液在0 ~1.5 V(vsLi/Li+)电压范围内的LSV 曲线。发现335C电解液从1.2 V 开始还原电流明显增加,表明335C电解液与金属锂负极之间发生了副反应;而凝胶电解质在这个电压范围内的还原电流明显减小,表明凝胶电解质中的335C 电解液与金属锂之间的副反应得到了一定程度的抑制。
为了进一步评估凝胶电解质对金属锂负极性能改善的效果,分别采用GPE 和335C 电解液组装金属锂对称电池,并对其界面稳定性进行对比。如图3(a)和图A4(a)所示,采用335C电解液所组装电池的界面阻抗静置15 d 后还在继续增加。而采用GPE 所组装的对称电池界面阻抗在第10 天就没有继续增加了,表明GPE 能够与金属锂负极更快地生成稳定的SEI,从而阻止GPE 中的335C 电解液与金属锂负极之间发生持续性的副反应。图3(b)展示了335C 电解液和GPE 分别组装的锂对称电池循环性能。采用GPE 所组装的对称电池在0.5 mA·cm-2的电流密度下稳定循环超过600 h。而在335C 电解液的对称电池中,循环500 h 后电压出现明显的上升,紧接着电池就失效了。图3(c)、(d)对比了这两组对称电池循环20周次后的金属锂负极表面形貌变化,相对于表面平整的新鲜电极来说[图A4(b)],在335C电解液中循环后的金属锂负极表面出现了蓬松的锂枝晶,而且还有明显被破坏的孔洞出现。而对于采用GPE 所组装的对称电池来说,电极表面相对平整,不同放大倍数下都没有出现明显的锂枝晶以及被损毁的表面形貌出现[图A4(c)、(d)],表面存在的一些凹痕是由于纤维素隔膜挤压所造成。
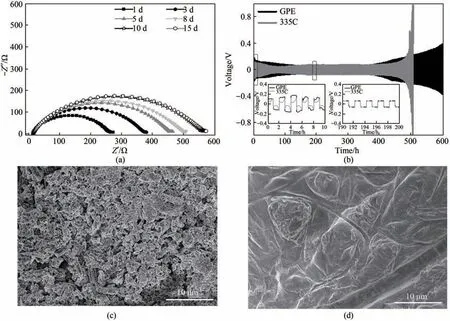
图3 (a)室温下Li|GPE|Li对称电池在不同静置时间的交流阻抗谱图;(b)采用GPE、335C电解液组装的Li|Li电池在电流密度为0.5 mA·cm-2,面容量为0.5 mAh·cm-2条件下的循环稳定性能图;(c)335C电解液及(d)GPE组装的锂负极经过20次循环后表面的SEM图Fig.3 (a)AC impedance spectra of Li|GPE|Li symmetrical cells for different time at room temperature;(b)Cyclic performance of Li symmetric cells with GPE,335C electrolyte at a current density of 0.5 mA·cm-2 with area capacity of 0.5 mAh·cm-2;Typical SEM images of the Li anode with(c)335Celectrolyte,and(d)GPE after 20 cycles
进一步采用原位聚合的方式组装了Li|GPE|LiFePO4电池,并在2.0 ~4.0 V电压范围内测定其在室温条件下的循环性能和倍率性能。图4(a)是Li|GPE|LiFePO4和Li|335C|LiFePO4电池在1 C 电流密度下的长周期充放电循环性能图。可以发现,335C 电解液所组装的电池仅循环100 周放电比容量就衰减到了110.8 mAh·g-1,而使用GPE 电解质的电池稳定循环超400 周后放电比容量仍能保持在118 mAh·g-1,其容量保持率在80%以上,且经过初始循环后,随后的电池库仑效率基本上达到99.5%以上。此外,在0.5C 的电流密度下(图A5),335C 电解液所组装的电池仅循环100 周容量保持率就下降到80.1%,而采用GPE所组装的电池充放电循环200周后的放电容量保持率仍有90.0%。相对于335C 电解液来说,基于GPE 所组装的电池表现出更好的循环稳定性能。图4(b)的DQ/DV 图显示Li|GPE|LiFePO4电池在充放电电压范围内只有Fe2+/Fe3+这一对氧化还原峰,表明充放电过程中没有其他额外的副反应。图4(c)展示了全电池的倍率性能,表明电池的放电容量随电流密度增大而有所下降。图4(d)为不同充放电倍率下的电压-比容量曲线,数据表明极化电压也随电流密度增加而变大。在0.1、0.5、1,2、3C下放电容量分别为161.2、139.7、123.0、98.0、78.7 mAh·g-1。上述的研究结果表明,相对于单纯使用335C电解液来说,原位聚合GPE 能够进一步提升LMBs 的电化学性能。
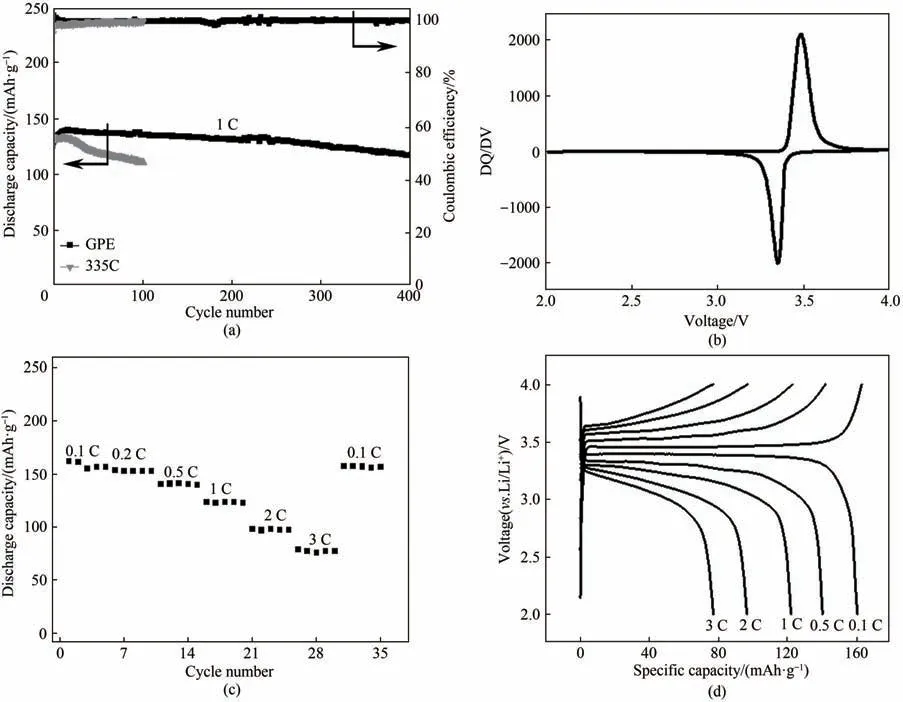
图4 (a)Li|LiFePO4电池在充放电电压为2.0 ~4.0 V以及电流密度为1 C条件下分别采用GPE和335C电解液的长期循环性能;(b)Li|GPE|LiFePO4电池放电曲线所对应的DQ/DV图;(c)Li|GPE|LiFePO4电池在不同电流密度下的倍率性能及其(d)对应的电压-比容量Fig.4 (a)Long-term cycling performance of Li|GPE|LiFePO4 cells(the cells were charged and discharged between 2.0—4.0 V at a current rate of 1 C);(b)DQ/DV diagram corresponding to charge and discharge curves of LMBs;(c)Rate performance of Li|GPE|LiFePO4 at various current rate;(d)Corresponding voltage profiles at various current rates
2.3 LMBs性能提升的原理机制
利用X 射线光电子能谱(XPS)分析对比了金属锂负极在两种电解质中循环50 周后的金属锂表面SEI 的成分。图A6 是基于GPE 电解质在金属锂表面的XPS 全谱图,可以明显观察到碳(C 1s)、氧(O 1s)、氟(F 1s)、磷(P 2p)和锂(Li 1s)的元素峰,这些元素主要来自于金属锂与凝胶电解质各组分之间反应的产物[33]。进一步对比两者表面SEI成分的F 1s 谱[图5(a)],其中使用335C 电解液的金属锂负极在684.5 eV 和686.6 eV 有两个主峰,分别为LiF 和LixPOyFz的信号峰[34]。而使用GPE 的金属锂负极,LiF 和LixPyFz两者产生的信号峰均有所偏移,分别位于684.9 eV和687.0 eV[35]。同时,发现两者的LiF 信号峰的面积在整个峰面积中的占比存在一定的差异,基于GPE的LiF 峰的面积占比约为80.6%,明显高于335C 电解液的占比(约45.6%)。上述结果表明,相较于使用335C电解液的金属锂负极而言,基于GPE的金属锂负极表面SEI 能够生成更多的LiF 物质。在Li 1s 谱图中[图5(b)],GPE 的金属锂表面存在三个峰信号,分 别 是 位 于56.1 eV 的LiF 峰[36]、位 于55.0 eV 的Li2CO3峰[37]以 及 位 于54.2 eV 的 酯 基 碳 酸 锂(ROCO2Li)峰[38]。对应的335C 电解液中金属锂表面的信号峰,分别位于56.6、55.5 和54.7 eV[39-40]。值得注意的是,GPE 中锂负极表面LiF 信号峰在整个峰面积中的占比约为10.0%,也明显高于335C 电解液(5.6%)。上述结果进一步表明,基于GPE 的金属锂负极表面SEI所含LiF 的量更高。此外,在F 1s谱中335C 电解液与GPE 的LiF 信号峰在整个峰面积占比之间的比值约为56%,接近于其在Li 1s 谱中的比值,两者表现出了较为一致的结果。因而,通过原位聚合生成凝胶电解质使得金属锂表面SEI中富含LiF,从而有效地提升了LMBs 的循环稳定性能。图5(c)展示了LiF 生成的反应路径示意图。反应过程①和过程②为335C 电解液中金属锂表面LiF 的主要来源,主要是通过LiPF6的分解平衡反应(过程①),另一部分则来自于PF5与金属锂(Li0)之间的反应(过程②)[20]。而在GPE 电解质中金属锂表面的LiF 来源得到了明显的增强,因为除了原本过程①和过程②所产生的LiF 外,由于THF 与PF5形成(THF)+(PF5)-的氧离子降低了电解液中PF5的浓度,这样就会促进分解反应平衡朝着正向移动,这个反应过程加剧了金属锂表面LiF 的生成。同时,由于引发聚合反应完成后,PF5会从聚合物末端释放出来,继续与金属锂反应生成LiF(过程③)。这些反应过程显著地增加了基于GPE 的金属锂负极表面SEI 中LiF 的含量。
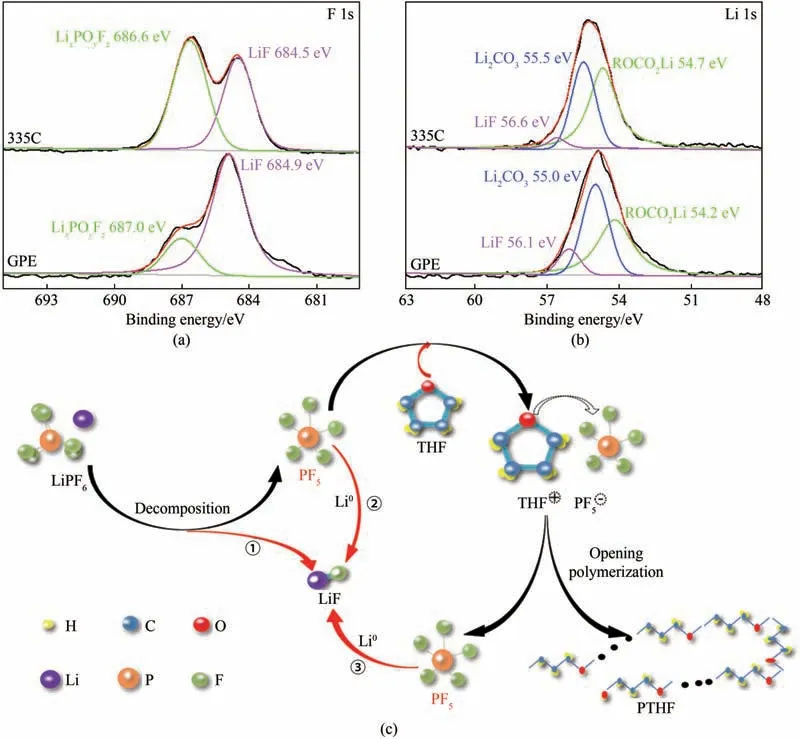
图5 GPE和335C电解液中循环50周后锂负极表面XPS的(a)F 1s谱图和(b)Li 1s谱图;(c)金属锂表面LiF物质生成反应路径示意图Fig.5 XPS spectra of Li metal(a)F 1s spectra,(b)Li 1s spectra in GPE and 335C electrolyte after 50 cycle;(c)Schematic diagram of LiF produce reaction
3 结 论
本工作主要基于LiPF6一物多用,将其作为GPE的导电锂盐、聚合引发剂以及构建金属锂负极SEI的氟源,以THF 为聚合物前体制备了一种高性能的凝胶电解质。这种GPE 具有较高的室温离子电导率(1.33 mS·cm-1)和宽的电化学稳定窗口[4.5 V(vsLi/Li+)]。采用原位聚合方式所组装的Li|GPE|LiFePO4电池,其循环稳定性获得了明显提升。这得益于凝胶聚合物电解质在开环聚合过程,能够促使LiPF6的分解反应平衡朝正向移动,从而增加了金属锂负极表面SEI 中LiF 的含量。这种富含LiF 的SEI 能够起到防止金属锂被335C 电解液持续性地腐蚀以及抑制锂枝晶的生长的作用。
附 录

图A1 THF与335C前体混合溶液质量比为1∶2(a),1∶3(b),1∶4(c)引发聚合后倒置状态的照片Fig.A1 The photograph of THF/335C precursor solution with mass ratios of 1∶2(a),1∶3(b),1∶4(c)after completion of polymerization

图A2 (a)Li|GPE|Li对称电池的阻抗拟合谱图;(b)Li|335C|Li对称电池的计时安培曲线(插图为对称电池极化前后的交流阻抗谱)Fig.A2 (a)The impedance spectra of Li|GPE|Li symmetric cell before and after chronoamperometry.(b)The chronoamperometry profile of Li|335C|Li symmetric cell(The inset displays the impedance spectra before and after chronoamperometry)
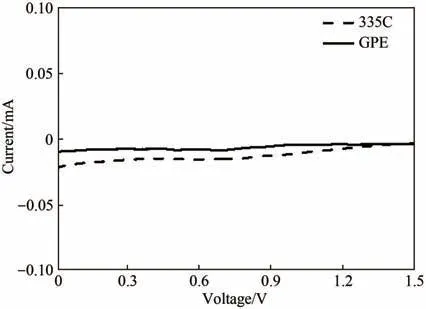
图A3 对应图2(d)中的GPE和335C电解液在LSV曲线中被还原分解区域内[0 ~1.5 V(vs Li/Li+)]放大图Fig.A3 The corresponding magnified views of two electrolytes decomposed at reduction reaction regions[0—1.5 V(vs Li/Li+)]in Fig.2(d)
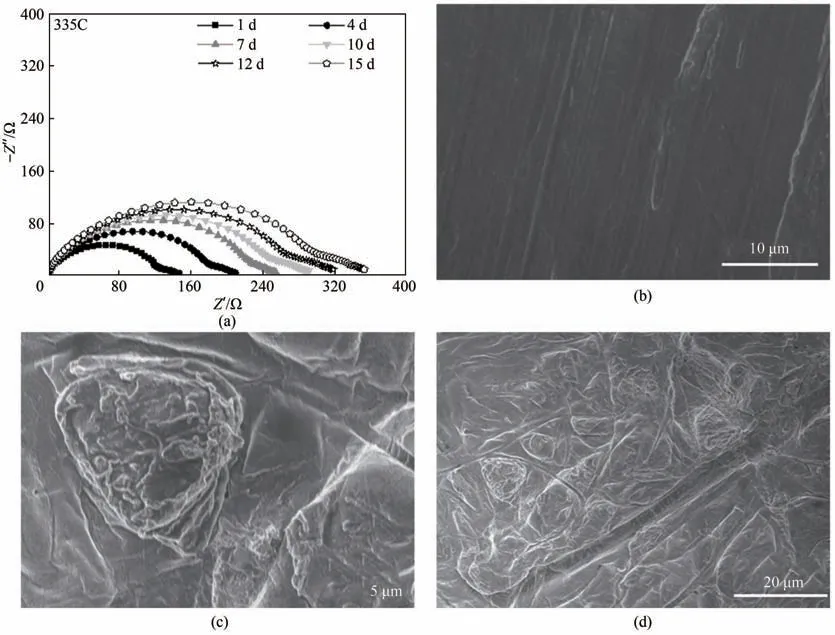
图A4 (a)室温下Li|335C|Li对称电池在不同静置时间的交流阻抗谱图;(b)新鲜锂负极表面的SEM图;(c),(d)GPE所组装的锂负极经过20次循环后金属锂表面在不同尺度下的SEM图Fig.A4 (a)AC impedance spectra of Li|335C|Li symmetrical cell for different time at room temperature;(b)Typical SEM images at the surface of fresh Li anode;(c),(d)Typical SEM images of the Li anode with GPE after 20th cycles under different scales

图A5 Li|LiFePO4电池在充放电电压为2.0 ~4.0 V以及电流密度为0.5 C条件下分别采用GPE和335C电解液的长期循环性能Fig.A5 Long-term cycling performance of Li|LiFePO4 cells with GPE or 335C electrolyte at a current rate of 0.5 C and voltage range of 2.0—4.0 V

图A6 Li|GPE|Li对称电池循环50周后金属锂表面的XPS谱图Fig.A6 XPS survey spectrum of lithium metal anode in GPE after 50 cycles
