2020年深圳地区4株冠状病毒HCoV-NL63基因组特征分析
2022-08-09阮嘉雯胡鹏威蒋潘虹鞠长燕刘楚云段永翔俞慕华
阮嘉雯,胡鹏威,蒋潘虹,鞠长燕,刘楚云,袁 梦,段永翔,陈 辉,俞慕华
人冠状病毒NL63(Human coronavirus NL63,HCoV-NL63)是呼吸道感染中常见的病毒之一,临床表现主要为病毒性感冒症状,在婴幼儿、老年人和免疫缺陷患者中可引起肺炎或中枢神经系统疾病[1-3]。Huang等[3]发现在台湾因肺炎住院病例中HCoV-NL63检出率为8.4%。2020年深圳市某看守所发生了一起由HCoV-NL63引起的呼吸道感染聚集性疫情,感染率为35.8%,远高于研究报道的HCoV-NL63感染率(1.0%~9.3%)[4-5]。在新冠肺炎疫情期间,本课题组所在的流感监测实验室进一步扩大对辖区内呼吸道感染疫情的病原学监测谱,发现HCoV-NL63检出率有所上升。现阶段我国HCoV-NL63尚未有健全的监测网络,国内的报道主要集中在HCoV-NL63在呼吸道感染病例中的检出率,但关于其全基因组序列信息、基因型特征及进化变异规律的报道仍较少。
HCoV-NL63为有包膜的单股正链RNA病毒,全长约27 538 bp,基因组结构从5′端到3′端依次为5′UTR-ORF1a/1b-S-ORF3-E-M-N-3′UTR[6]。棘突蛋白(Spike protein,S)和膜蛋白(Membrane protein, M)是HCoV-NL63两个重要的糖蛋白。研究发现,M蛋白在HCoV-NL63感染早期扮演重要的角色,S蛋白与M蛋白的协同作用是病毒与受体细胞结合并介导膜融合的先决条件[7-9]。由于HCoV-NL63和可引起重症肺炎的冠状病毒SARS-CoV、SARS-CoV-2均以血管紧张素转换酶2(Angiotensin-converting enzyme 2, ACE2)为受体,具有诱发严重下呼吸道感染的风险[10-11],因此,监测HCoV-NL63遗传变异和致病性仍具有重要的现实意义。
本研究对新冠疫情期间深圳市南山区呼吸道感染聚集性疫情[4]和散发疫情中获得的4株HCoV-NL63核酸阳性样本进行全基因组测序,并结合GenBank数据库中其他国家和地区HCoV-NL63流行株序列信息,对HCoV-NL63基因特征及S蛋白、M蛋白的N-糖基化位点进行分析比较,在一定程度上丰富了我国HCoV-NL63分子流行病学监测数据,同时也为HCoV-NL63的诊断治疗及防控提供数据支持。
1 材料与方法
1.1 标本来源 本研究中4份HCoV-NL63核酸检测阳性标本分别来自于深圳市南山区一起呼吸道感染聚集性疫情[4]和学校呼吸道感染散发疫情的咽拭子标本。
1.2 病毒检测和基因测序 通过天隆核酸提取仪(西安天隆科技有限公司)提取咽拭子标本中病毒总RNA,利用实时荧光定量PCR法(RT-PCR)检测HCoV-NL63,试剂为15项呼吸道病原体快速检测试剂盒(联合医学)。HCoV-NL63核酸检测阳性样品由深圳华大基因科技有限公司进行二代测序。
1.3 序列分析 在GenBank数据库中筛选并下载1983—2018年全球HCoV-NL63流行株。进一步对比剔除相同的HCoV-NL63核苷酸序列后,利用MEGA 6.0软件对下载的具有代表性的55条HCoV-NL63全基因组序列构建全基因组亲缘关系进化树。采用最大似然法(maximum likelihood,ML),选择计算得出的GTR+G替换模型构建系统发生树,并使用p-distance估计遗传距离,bootstrap值选择1000次引导重复测试准确性,bootstrap值>60%认为分组具有统计学意义。采用Bioedit软件计算本研究获得的HCoV-NL63毒株序列与GenBank数据库中下载的其他国家和地区55条参考株全基因组序列氨基酸熵值,分析其变异情况。同源性比较及S蛋白、M蛋白氨基酸位点突变分析通过MEGA 6.0软件完成。氨基酸突变位点分析选用B基因型参比株AY518894.1。
2 结 果
2.1 基因组序列信息 本研究通过二代测序技术获得了4株HCoV-NL63全基因组序列(表1),推测其基因组结构从5′端到3′端依次为5′UTR、ORF1a、ORF1b、S、ORF3、E、M、N、3′UTR,基因组两端分别含有转录非翻译区(UTR)。
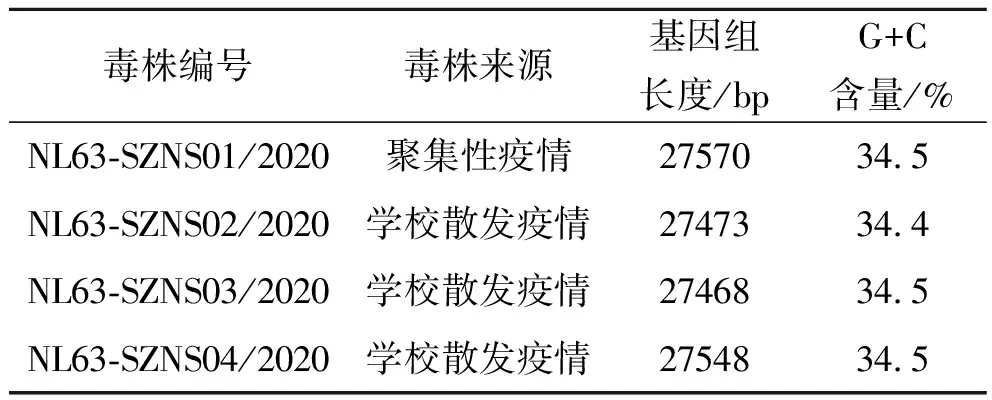
表1 4株HCoV-NL63毒株信息
2.2 进化树分析 将本研究中获得的4株序列与GenBank数据库中下载1983—2018年全球具有代表性的HCoV-NL63 S基因序列和全基因组序列构建进化树分析。S基因的进化树分析结果显示(图1A),HCoV-NL63毒株划分为A、B、C 3个基因型,其中本研究获取的4株毒株均属于B基因型B2基因亚型,在进化树上处于同一分支,与2018年广州发现的MK334046.1、MK334047.1株亲缘性关系较近。S基因的进化树跟全基因组进化树(图1B)结果相似。

▲指本研究获得的毒株。A: 以S基因序列构建进化树; B: 以全基因组序列构建的进化树。
2.3 序列同源性分析 将本研究中获得的4株HCoV-NL63毒株和6株参考株的全序列进行同源性分析,结果见表2。本研究获得的4株毒株间的核苷酸(氨基酸)同源性为99.80%~99.98%(99.64%~99.93%),与2018年广州发现的B基因型MK334046.1同源性为99.79%~99.90%(99.68%~99.84%),与B型参考株AY518894同源性为98.52%~98.59%(97.81%~97.90%);与原型株NC 005831.2同源性为97.64%~97.70%(96.85%~96.92%)。
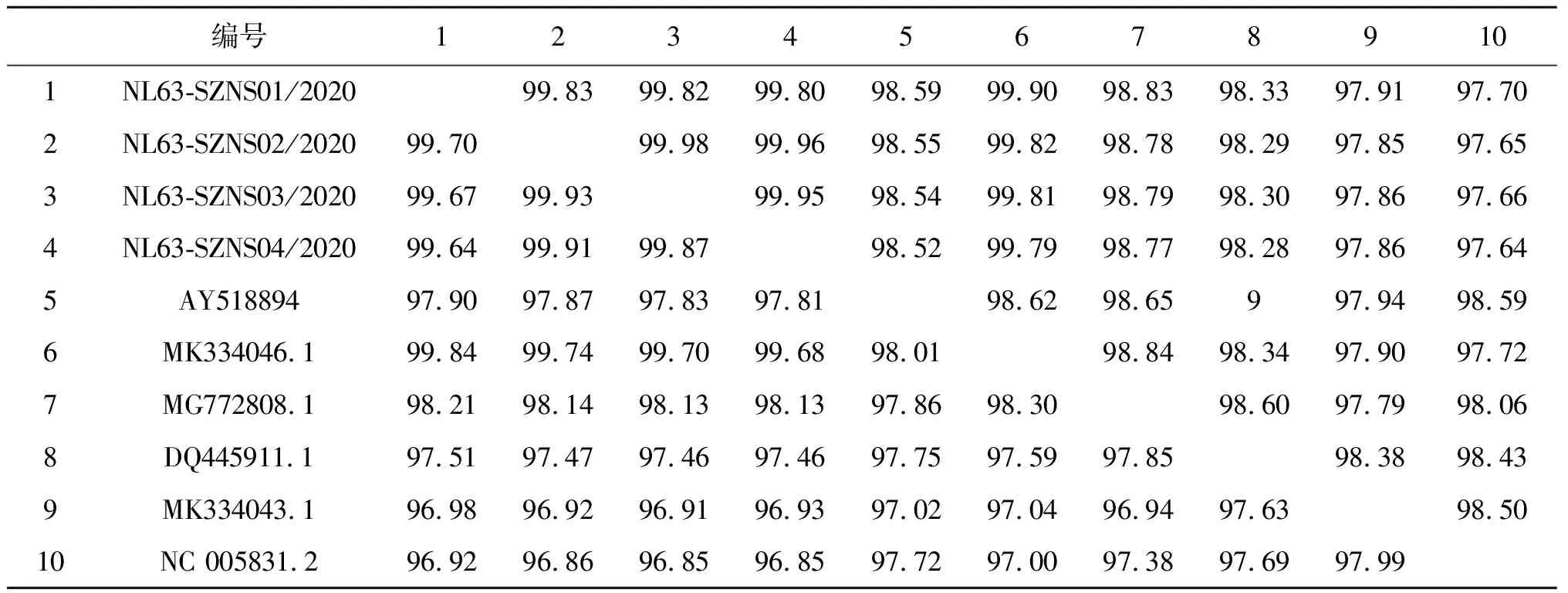
表2 2020年深圳市南山区4株HCoV-NL63毒株与参考株全序列同源性分析
2.4 全基因组基因多态性分析 采用BioEdit软件(v7.9.0)Entropy[H(X)]plot计算从NCBI数据库中下载的55条HCoV-NL63流行株序列及本研究获得的毒株序列各蛋白编码区氨基酸位点的熵值。由图2分析发现,变异性较大的蛋白编码区分别为ORF1ab蛋白、S蛋白和M蛋白。全基因组中变异熵值≥0.6的易突变氨基酸位点共有193个,其中ORF1ab蛋白编码区最多,有90个,占蛋白编码区总变异位点数46.6%;其次为S蛋白,有83个(43%)易突变位点;M蛋白、N蛋白、E蛋白分别有9、5、2个位点。
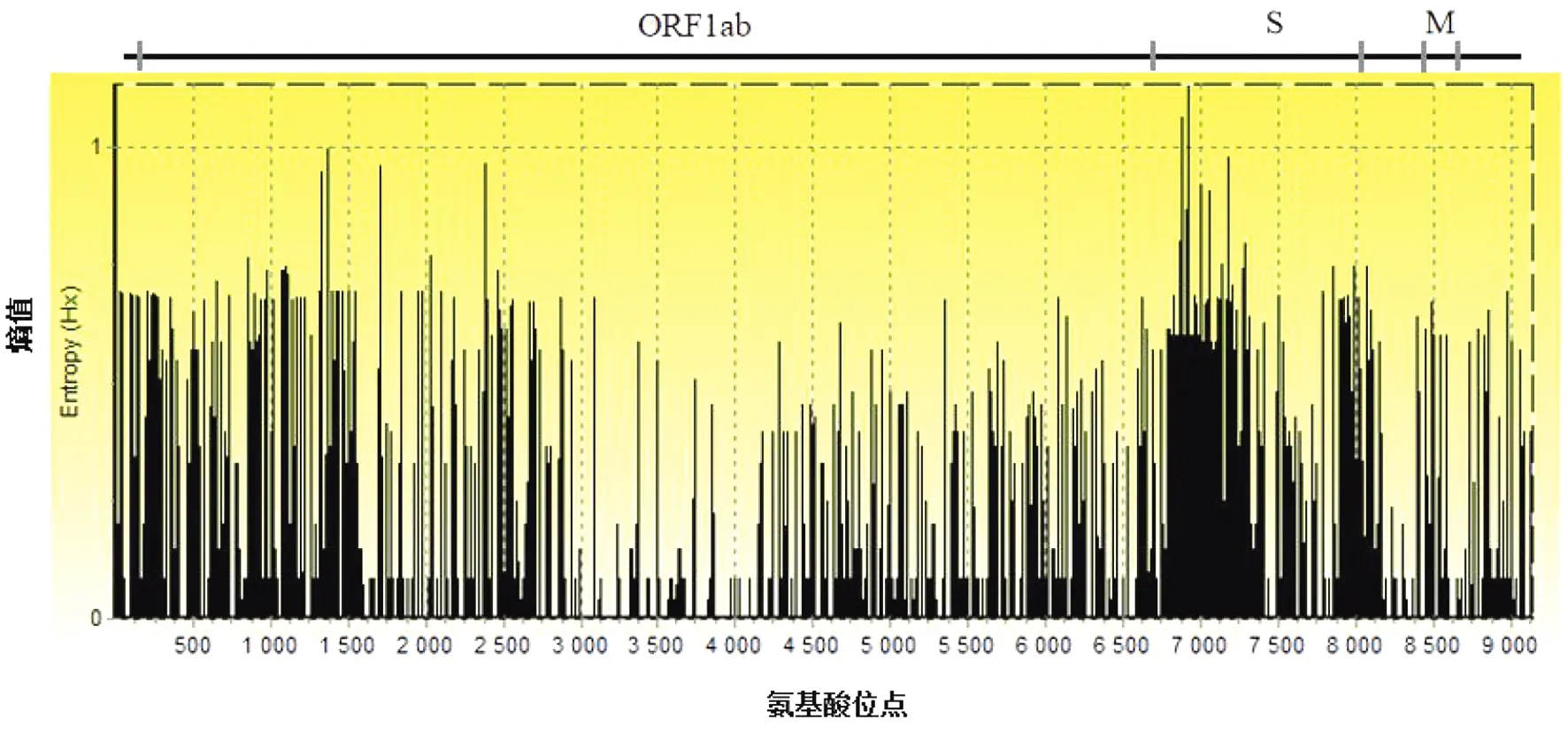
图2 全基因组各个蛋白质编码区氨基酸变异
将获得的4株HCoV-NL63序列和B型的参考株进行S蛋白、M蛋白氨基酸位点差异分析。结果发现,S蛋白有48个单氨基酸多态性位点(图3),其中在本研究从学校疫情获得的3株毒株(NL63-SZNS02/2020、NL63-SZNS03/2020、NL63-SZNS04/2020)的S1蛋白区第196位均发生了亮氨酸→苯丙氨酸的突变;聚集性疫情NL63-SZNS01/2020毒株S2蛋白区第946位发生了氨基酸替换(A946S)。M蛋白有8个单氨基酸多态性位点(图4)。
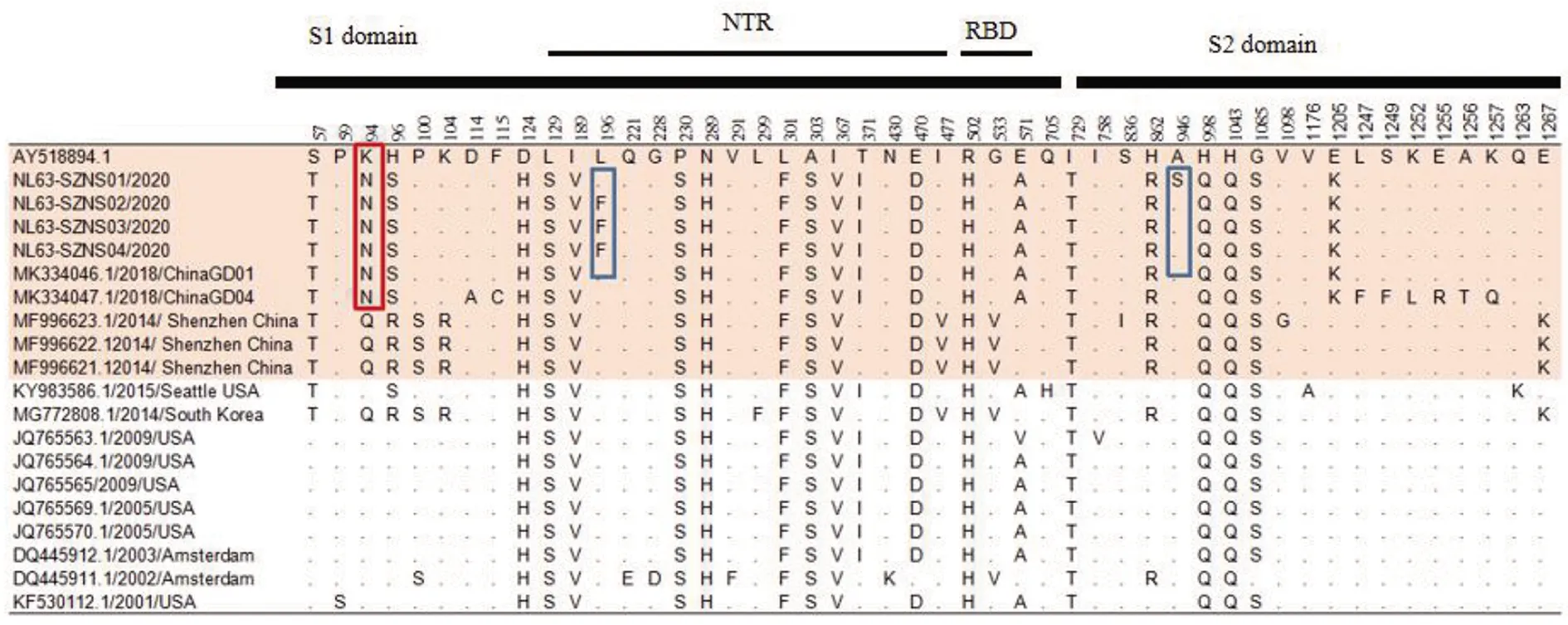
图3 获得的4株HCoV-NL63毒株与B型参考株S蛋白氨基酸差异位点比较
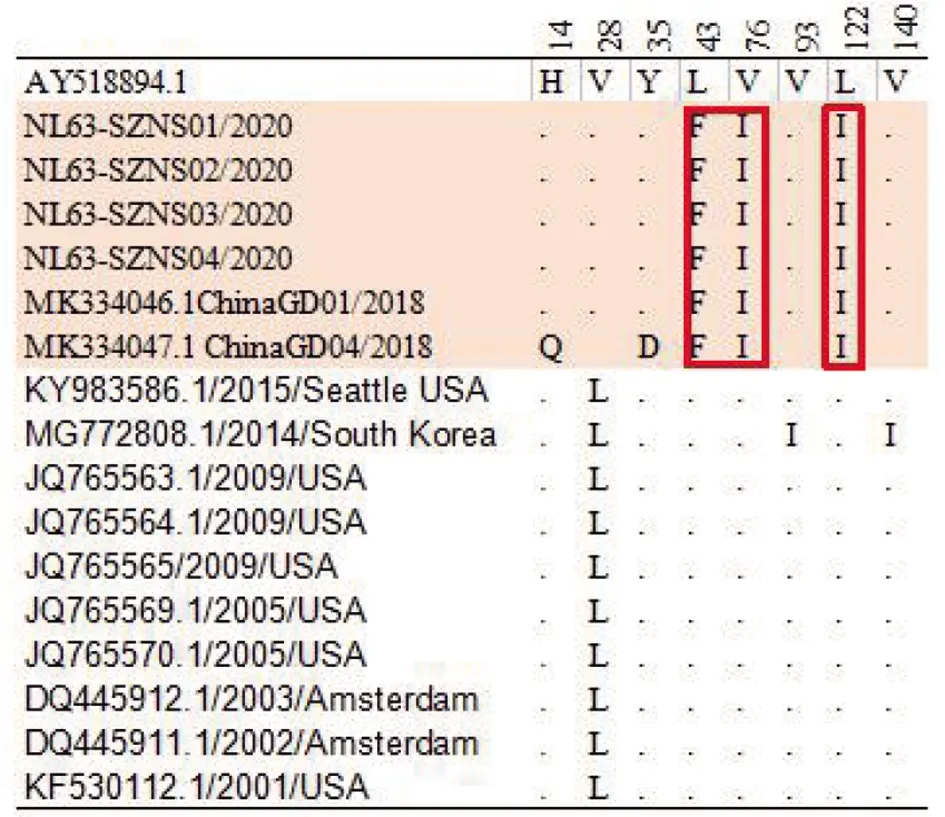
图4 获得的4株HCoV-NL63毒株与B型参考株M蛋白氨基酸差异位点比较
2.5 N-糖基化位点分析 通过NetNGlye 1.0在线预测了获得的HCoV-NL63毒株S蛋白和M蛋白的N-糖基化位点。结果显示,S蛋白有10个糖基化位点(阈值>0.5),分别为94 NISL、117 NVST、153 NATR、299 NFSS、424 NVSA、504 NISL、842 NVTS、850 NLSS、1216 NKTL、1245 NLSS;M蛋白的N-糖基化位点有2个(阈值>0.5),分别为3NSSV、19NFSW。
3 讨 论
HCoV-NL63是目前已知的唯一能与SARS-CoV和SARS-CoV-2的受体ACE2结合,从而侵染细胞并在胞内进行病毒复制的α属冠状病毒[12]。Van等[13-14]发现在几乎所有8岁以上儿童血清中均能检测到针对HCoV-NL63的有效中和抗体,表明HCoV-NL63感染在婴幼儿时期非常普遍。虽然HCoV-NL63临床表现主要为轻微的呼吸道感染症状,但随着病毒的持续传播和进化,其致病性也会发生变化。2018年Wang等[15]在广州发现HCoV-NL63病毒的B基因型和C3亚型可诱发严重的下呼吸道感染症状,推测S蛋白RBD区域I507L位点的突变可能会加强HCoV-NL63的传播力和致病力。因此,监测HCoV-NL63基因遗传变异特性,对评估HCoV-NL63流行风险和制定相应的防控措施有着重要的作用。
本研究从聚集性疫情和散发疫情HCoV-NL63核酸检测阳性标本中,利用二代测序技术获得了4株不同来源的HCoV-NL63全基因组序列,并进行基因型鉴定和基因特征分析。结果显示本研究中4株不同来源的HCoV-NL63毒株序列高度相似,并且与广州肺炎病人的MK334046.1、MK334047.1在基因进化树形成一个独立的进化分支,提示来源于同一传播链;对已报道深圳地区流行的HCoV-NL63毒株S蛋白进行进化分析,此次在南山区疫情中获得的4株毒株与2014年深圳市流行的HCoV-NL63毒株被划分为不同的基因亚型,推测深圳地区HCoV-NL63可能存在不同的传播链,引起聚集性疫情和散发疫情的HCoV-NL63病毒源自广州毒株的可能性大。由于目前已知的深圳地区HCoV-NL63毒株序列的时间空间覆盖不足,尚不能全面的解释深圳地区HCoV-NL63毒株变异变迁情况,后续仍需收集更多的流行毒株序列信息。
对熵值结果分析发现,变异在基因组广泛存在,结构蛋白中S蛋白氨基酸变异最大,因此S蛋白经常作为分子流行病学研究病毒传播链的靶基因片段。前期研究发现,S蛋白与M蛋白在病毒识别受体细胞、侵染细胞并在胞内进行病毒复制的过程中起着重要的作用[7]。S蛋白是冠状病毒中最大的跨膜糖基化蛋白,胞外域可以被分成介导受体结合的S1和介导膜融合的S2两个蛋白亚基。S1亚基是主要的功能区,包含两个功能域:N端结构区(N-terminal region,NTR)和受体结合区(Receptor binding domain,RBD)(476~616 aa)[15-16]。目前认为N端结构区是S蛋白中基因变异频率最高的区域,含有多个潜在的N-糖基化位点[17]。S2亚基进一步使病毒包膜和细胞膜重新排列,拉近病毒膜和细胞膜间的距离,启动膜融合的发生,介导病毒侵入细胞[18]。与B型参考株相比,本研究获得的4株HCoV-NL63株的S蛋白中发现2处氨基酸位点变异:位于散发疫情毒株S1蛋白NTR区L196F和位于聚集性疫情毒株S2蛋白A946S。A946S突变是否会影响聚集性疫情毒株与细胞膜融合过程从而引起感染率升高,仍需深入研究。进一步对GenBank数据库中HCoV-NL63病毒B基因型流行株序列分析发现,2018年来中国广东地区B基因型流行株中S1亚基94氨基酸发生了替换(K94N),增加了94位N-糖基化位点94 NISL。S1蛋白区域的N-糖基化位点增加可能会引起氨基酸空间构象的改变,可能会在一定程度上帮助病毒逃逸宿主中和抗体识别[19-20]。本研究获得的广东地区HCoV-NL63 B基因型流行株中N-糖基化位点增加对病毒抗原性及机体免疫的影响还有待进一步的探讨。
M蛋白通过与宿主细胞表面硫酸肝素糖蛋白(HSPGs)结合,介导病毒附着于宿主细胞,在HCoV-NL63早期感染阶段起着重要的作用。M蛋白包含一个氨基端膜外区、一个跨膜区和一个羧基端膜内区,氨基端153~226 aa区被认为是HSPG结合位点[7, 21-22]。对GenBanK数据库中HCoV-NL63病毒B基因型流行株M蛋白分析发现,本研究发现M蛋白153~226 aa区域较为保守,与B型参比株相比均没有发生氨基酸替换;在广东流行株中均发现3个特异性的氨基酸位点变异(L43F、V76I、L122I),这些氨基酸位点的突变对HCoV-NL63的致病力和传播能力的影响仍有待研究。
本研究对深圳地区聚集性疫情和散发疫情获得的4株HCoV-NL63进行全基因组测序和基因特征分析,初步分析了与病毒入侵相关的S蛋白、M蛋白的氨基酸变异情况及潜在的N-糖基化位点,在一定程度上丰富了我国HCoV-NL63基因特征信息,为相关的防控工作提供生物学依据。
利益冲突:无
引用本文格式:阮嘉雯,胡鹏威,蒋潘虹,等.2020年深圳地区4株冠状病毒HCoV-NL63基因组特征分析[J].中国人兽共患病学报,2022,38(1):35-41.DOI:10.3969/j.issn.1002-2694.2021.00.178
