钴、氮、硼掺杂氧化石墨烯催化剂的制备及催化臭氧氧化性能
2022-08-03郭子毅
张 远,郭子毅,王 栋
(大连理工大学 环境学院,辽宁 大连 116024)
催化臭氧氧化技术是一种废水深度处理方法,近年来引起了人们的广泛关注,而单独臭氧由于氧化能力低于羟基自由基且具有选择性,限制了其应用范围。相比单独臭氧氧化,催化臭氧氧化能快速生成羟基自由基等活性基团,氧化分解水中一些持久性和高毒性的污染物。石墨烯材料催化臭氧氧化水中污染物是水处理研究的焦点之一,石墨烯材料可以通过促进臭氧分解和活性基团的产生提高污染物的去除率。设计具有增强臭氧转化为活性基团性能的石墨烯材料,选择性地调节表面活性中心的类型和密度,具有重要研究价值。
金属和非金属双原子或多原子掺杂的石墨烯材料通常对污染物有更好的降解效果。然而,在多原子掺杂催化剂的制备过程中,金属和非金属元素掺杂比的不同会影响催化剂成型的结构和理化性质,进而对最终水中污染物的去除效果产生影响。因此,探究不同比例的金属和非金属元素掺杂石墨烯材料的性能对催化臭氧氧化降解水中污染物具有重要意义。
本研究通过调控金属钴与非金属氮、硼元素的相对含量,采用水热—高温煅烧法制备了不同钴负载量的钴、氮、硼掺杂氧化石墨烯(Co/B-NGO)催化剂,以对氯苯甲酸为探针化合物进行了自由基探针实验,以布洛芬为目标污染物进行了催化臭氧氧化实验,并以草酸为氧化中间产物进行了自由基诱导矿化实验,为进一步扩大该类催化剂的应用提供参考。
1 实验部分
1.1 试剂和材料
氧化石墨烯(GO):片径0.5~5.0 μm,厚度0.8~1.2 nm,江苏先丰纳米材料科技有限公司。
草酸、布洛芬、对氯苯甲酸、叔丁醇(TBA)、六水合硝酸钴、2-甲基咪唑、甲醇、硼酸、乙醇:均为分析纯。实验用水:超纯水。
1.2 催化剂的制备
将900 mg的2-甲基咪唑和380 mg的硼酸溶于28 mL甲醇中,然后加入7 mL含GO的甲醇悬浮液(5 g/L),超声处理1.5 h。加入六水合硝酸钴的甲醇溶液35 mL,继续搅拌0.5 h。混合物转移至四氟乙烯内胆并置于不锈钢高压釜中,于150 ℃加热4 h。加热完成后,待高压釜冷却至室温,取出溶液抽滤,沉淀物用乙醇和超纯水洗涤后真空干燥。将洗涤、干燥后的沉淀物置于通氮气的管式炉中,高温煅烧2 h。
1.3 催化臭氧氧化实验
本实验采用内径0.1 m、有效容积0.25 L的半间歇式密闭玻璃反应器进行反应。调节气体流量器并利用多孔曝气头将稳流后的臭氧送入反应器底部,尾气经质量分数为2%的碘化钾溶液吸收。反应器全程被放置在(25.0±0.5) ℃的恒温房中,使用磁力搅拌器将固体催化剂均匀分散于溶液中,臭氧流量控制在(0.50±0.01) L/min。
实验开始前配制50 mg/L有机底物溶液100 mL并装入反应器中,接着开启磁力搅拌器和臭氧发生器,调节臭氧气体流量,待气流稳定后加入0.025 g催化剂,打开曝气阀并开始计时,整个实验在恒温(323 K)环境中进行,臭氧质量浓度10 g/m。实验过程中每隔一定时间用取样针从反应器中取样2 mL,并立即经高纯氮气吹扫样品中的残留臭氧。样品中残留的微量催化剂经0.22 μm有机滤膜过滤后,通过高效液相色谱仪测定目标化合物浓度,进行三次平行实验,取平均值。
1.4 分析方法
采用X射线衍射仪(SmartLab型,日本Rigaku公司)分析催化剂表面化学成分及晶型结构特征;采用场发射扫描电子显微镜(SU8010型,日本日立公司)对催化剂表面的微观形貌进行观察;采用X射线电子能谱仪(ESCALAB XI+型,英国Thermo公司)分析催化剂组成元素的化合价状态;采用傅里叶变换红外光谱仪(EQUINOX55型,德国Broker公司)分析催化剂的表面官能团。
采用高效液相色谱仪(Agilent 1100型,美国Agilent公司)测定草酸、对氯苯甲酸、布洛芬的浓度,色谱柱为Yilite C18 5 μm柱(4.6 mm × 250 mm)。
2 结果与讨论
2.1 催化剂的表征结果
2.1.1 SEM
不同钴负载量(制备催化剂时钴元素的质量分数)催化剂(煅烧温度500 ℃)的SEM照片如图1所示。催化剂整体呈现表面褶皱的无规则片层结构。随着钴负载量的增加,催化剂表面形成颗粒的覆盖面积也增大并均匀分散。8%负载量催化剂的放大SEM照片揭示了金属团簇的形成,且金属团簇的粒径小于2 μm并呈现镂空多孔形态。

图1 不同钴负载量催化剂的SEM照片
2.1.2 XRD
催化剂的XRD谱图见图2。如图2a所示:在700 ℃的煅烧条件下,未掺杂钴的催化剂的XRD谱图与BCN标准谱图(PDF#53-0447)峰型拟合良好,且石墨峰(26.2°)明显,表明非金属元素硼和氮成功掺杂到石墨烯中;随着钴负载量的增加,GO表面被掺杂原子及其氧化物所覆盖,石墨峰逐渐减弱,在8%钴负载量催化剂的XRD谱图中几乎消失。如图2b所示,以8%钴负载量催化剂为实验对象,300 ℃煅烧后金属钴在催化剂中以CoCoO(PDF#80-1532)双价态存在,500 ℃煅烧后以CoO(PDF#75-0533)单价态存在,700 ℃煅烧后以Co(PDF#15-0806)单质存在,未形成其他钴氧化物。原因可能是随着煅烧温度的升高,钴先在低中温被氧化形成不同价态的钴氧化物,而在高温中钴氧化物被石墨烯中的碳原子还原,形成钴单质。
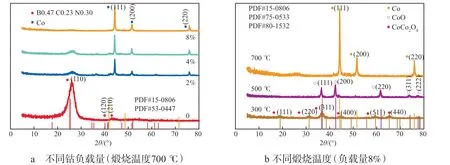
图2 催化剂的XRD谱图
2.1.3 FTIR
不同钴负载量催化剂(煅烧温度500 ℃)的FTIR谱图如图3所示。除未掺杂钴的催化剂外,另外3个不同钴负载量催化剂的FTIR谱图未见明显的表面官能团变化。3 430 cm处的宽峰为O—H键伸缩振动特征峰,且峰强随钴负载量的增大而增强,原因可能是在高温煅烧过程中催化剂表面的钴氧化物被碳还原,有利于O—H键的形成。2 920 cm和2 850 cm处的峰是亚甲基(—CH—)的伸缩振动特征峰。1 635 cm处的峰为共轭的链烯烃(>C=C<)官能团的特征吸收峰,该特征峰的出现可能是由于非金属元素氮、硼掺杂改变了石墨烯原有的电子排列,使共轭体系中的电子云密度趋于平均化,导致原来的双键略有伸长,吸收频率向低波数方向移动。1 075 cm处的峰为C—O键伸缩振动特征峰。640~570 cm处的峰为金属钴的特征峰。
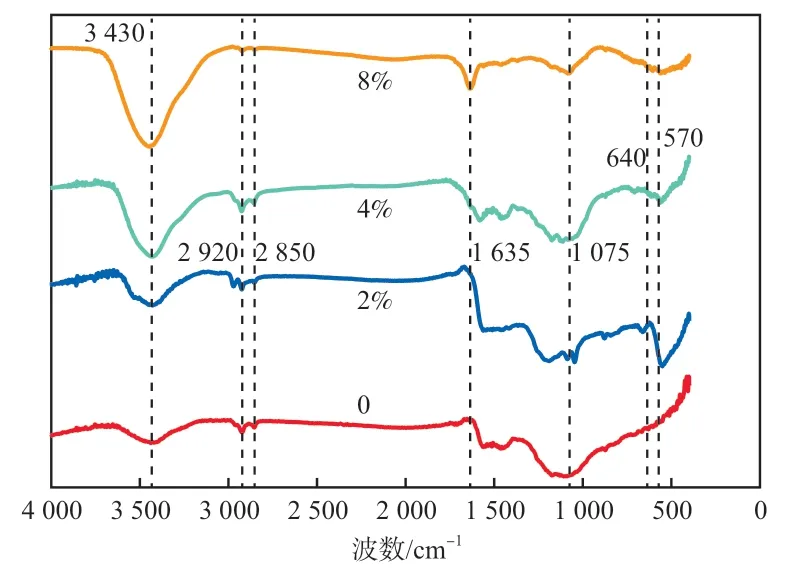
图3 不同钴负载量催化剂的FTIR谱图
2.1.4 XPS
催化剂(钴负载量8%、煅烧温度500 ℃)的XPS谱图见图4。催化剂的C 1s谱图分别在284.6 eV出现C—C sp特征峰,在285.3 eV出现C—N/C—C sp特征峰,在286.1 ev出现C—O特征峰以及在283.6 eV出现C—B特征峰,C—B特征峰的出现进一步证实了硼原子在催化剂载体上的掺杂。催化剂中Co 2p的谱图呈现的不同特征峰分别对应Co2p(778.3 eV)、Co2p(780.0 eV)、Co—N2p(781.3 eV)、Co2p(793.3 eV)、Co—N2p(796.3 eV)和Co2p(796.5 eV),其中Co—N特征峰结合C 1s的C—N特征峰,间接证实了Co—N—C结构的形成。N 1s谱图中分别在398.4,399.0,399.8,401.1 eV拟合出4个特征峰,分别对应吡啶氮、B—N、吡咯氮和石墨氮。催化剂的O 1s谱图显示,氧元素大部分以C—O—C/—COOH(531.45 eV)的形态存在于催化剂中,另外少量与金属钴结合形成Co—O(529.6 eV),其余的以C=O(533.6 eV)形态存在。
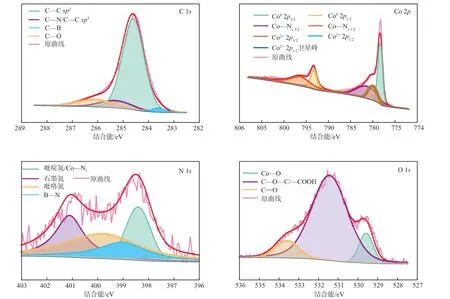
图4 催化剂的XPS谱图
2.2 目标有机物的催化臭氧氧化
不同催化剂催化臭氧氧化的布洛芬去除效果如图5所示。图5a显示出单独臭氧氧化(即无催化剂)体系的反应速率较小,在体系中加入不同钴负载量催化剂后反应速率均有提升,其排序为:无催化剂< 4% < 0 ≈ 2% < 8%。从总体趋势来看,随着催化剂钴负载量的提高,催化活性未见相应增高,原因可能是催化剂的表面活性位点数量与孔隙率之间存在竞争关系。在制备催化剂的过程中,不同的煅烧温度同样会影响催化剂的活性,如图5b所示。选取最佳钴负载量(8%),分别在300 ℃、500 ℃和700 ℃煅烧制备催化剂,并进行催化臭氧氧化布洛芬实验,最终效果为:500 ℃> 700 ℃> 300 ℃,其中500 ℃煅烧制备的催化剂反应10 min的布洛芬去除率即可达99%。原因可能是煅烧温度对催化剂表面钴的存在价态有影响,进而导致催化活性不同。这与图2催化剂的XRD谱图分析结果相符。
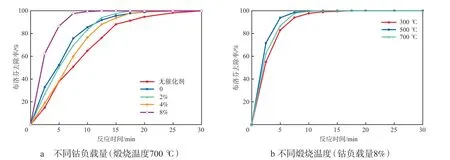
图5 不同催化剂催化臭氧氧化的布洛芬去除效果
草酸是污染物降解过程中一种常见的中间产物,单独与臭氧很难发生降解反应(反应速率常数0.04 mol/(L·s)),因此将草酸作为考察氧化中间产物。如图6所示:单独臭氧40 min的草酸去除率仅为7.7%;在加入催化剂(煅烧温度500 ℃)的体系里,草酸的降解速率和降解程度均有明显提升。其中,添加8%钴负载量催化剂降解草酸的效果最为显著,在10 min里草酸去除率达91.3%,远高于单独臭氧氧化。Co/B-N-GO催化剂的加入增加了催化体系活性反应位点的数量,并通过GO载体提高了电子传递效率和反应接触面积,增强了体系的氧化能力,说明通过催化剂表面介导的催化作用可以产生更多的自由基。证明Co/B-N-GO催化剂对自由基诱导的矿化反应表现出很高的活性,有利于提高难降解有机物的矿化程度。
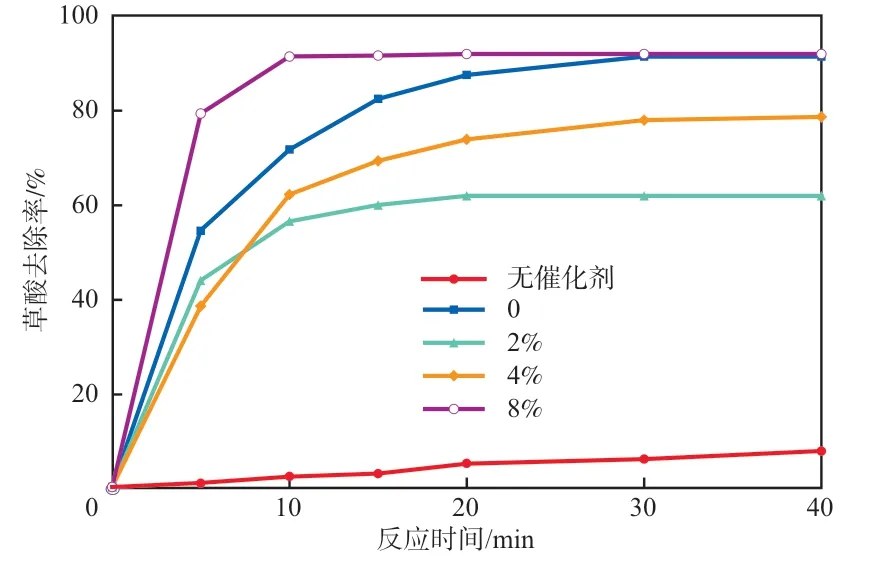
图6 不同钴负载量催化剂的草酸去除效果
对氯苯甲酸由于与·OH反应灵敏且快速(反应速率常数5×10mol/(L·s)),与臭氧分子反应慢(反应速率常数小于0.15 mol/(L·s)),因此,在催化臭氧氧化体系中常被用来检测·OH。如图7所示,无论是单独臭氧还是催化臭氧氧化体系(催化剂钴负载量8%、煅烧温度500 ℃),对氯苯甲酸均在5 min内快速被降解,两者无显著差别。在两体系中分别加入自由基淬灭剂TBA探究体系中对氯苯甲酸遵循的反应途径。臭氧可以在水中自分解产生·OH,TBA的加入抑制了臭氧对对氯苯甲酸的降解。对于催化臭氧氧化体系,TBA对对氯苯甲酸的降解抑制作用没有单独臭氧体系明显,加之由图可见催化剂对对氯苯甲酸吸附作用微弱,推测可能是催化剂表面区域产生了局域性催化效应,加大了·OH的暴露,对氯苯甲酸与催化剂表面接触时被快速降解。TBA可以快速淬灭水中的·OH,但不能淬灭催化剂表面产生的自由基,这与实验结果相符。
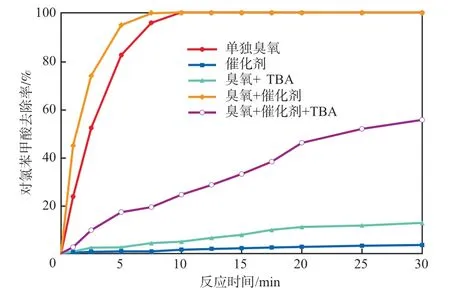
图7 不同工艺条件下对氯苯甲酸的去除效果
2.3 反应动力学
为了进一步探讨催化剂的催化作用,建立了动力学模型表达污染物的去除效果。通过比较不同催化条件下的表观一级反应动力学,可以更直观明了地理解非均相催化臭氧化反应中不同催化材料对有机物降解的能力。非均相催化臭氧氧化体系一般符合表观一级动力学方程,故本实验的反应方程可写为式(1)。结合实验中·OH在催化反应中起主导作用,可将式(1)简化为式(2),进而得到式(3)。
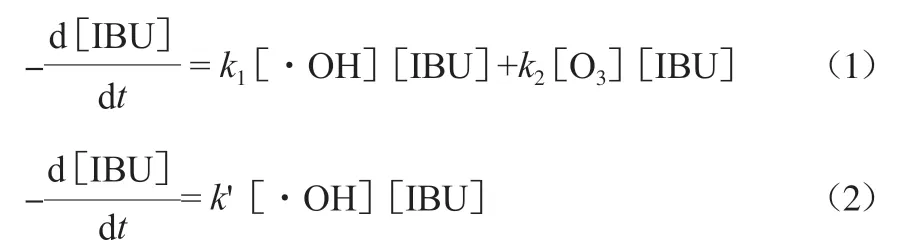

式中:t为反应时间,min;k和k分别为·OH和O与IBU(布洛芬)的反应速率常数,min;k′ 为修正后的反应速率常数,min;k为表观一级反应速率常数,min;b为实验误差(无误差时b=0);c和c分别为t时刻和初始时的IBU浓度。
按式(3)对实验数据进行拟合,结果表明,不同催化剂的催化臭氧氧化过程均很好地符合表观一级反应动力学方程,R均达0.965 0及以上,具体参数如表1所示。
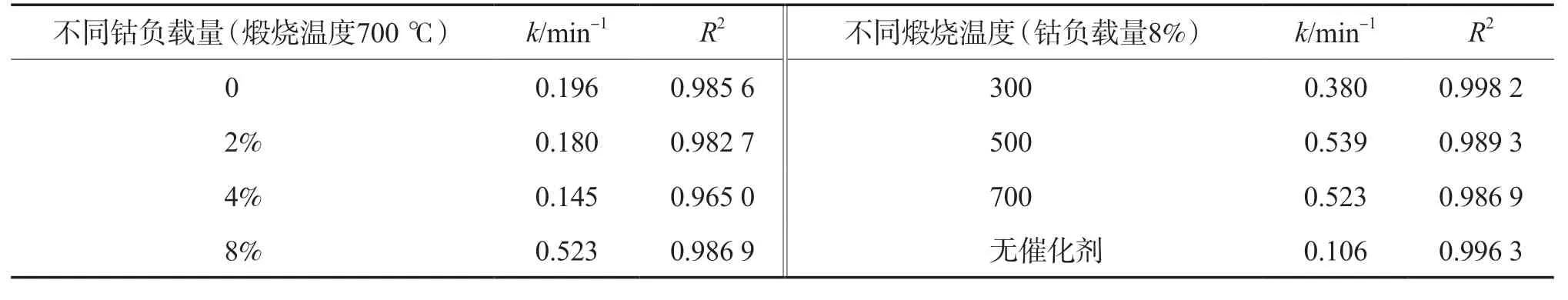
表1 不同催化剂的表观一级动力学拟合参数
k值可作为评价催化剂活性的指标,较大的k值表示与目标污染物有较高的反应亲和力。不同钴负载量和煅烧温度的催化剂在催化降解布洛芬时k值有所差异,其中当钴负载量为8%且煅烧温度为500 ℃时,催化剂的催化效果最佳,k值为0.539,接近单独臭氧体系(0.106)的五倍。该结果与布洛芬催化臭氧氧化实验的研究结果一致。
2.4 催化臭氧氧化机理推测
当催化剂投入通有臭氧的溶液中后,催化剂表面首先与臭氧分子接触,催化剂表面的二价钴被臭氧氧化生成三价钴,同时在催化剂表面生成·OH(式(4))。另一方面,由于催化剂载体为多原子掺杂的GO,含有大量的π电子,在π电子的作用下,反应生成重要的中间产物·O和OH(式(5)~(7))。在·O和OH的参与下,三价金属钴被还原为二价金属钴((式(8)~(9))。
2.5 催化剂的再生性能
图8为催化剂高温再生(85 ℃煅烧2 h)后的重复实验结果。5次再生重复性实验发现,催化剂仍保持了较好的催化臭氧氧化草酸的性能。首次使用催化剂40 min可去除水中91.3%的草酸,第2~5次去除率分别为89.3%,87.2%,85.2%,84.1%,第5次活性仍达到了初次使用的92.1%。5次重复利用催化剂后,降解草酸的反应速率常数为0.048 2 min,与首次使用时的0.050 8 min接近。随着重复利用次数的增加,催化剂对草酸的去除率略有下降,原因可能是在长时间的搅拌条件下催化剂之间相互摩擦碰撞,钴含量及形貌发生了微小改变。此外,催化剂不断受到臭氧分子表面冲击,可能会导致催化剂表面部分反应活性位点的缺失或者被其他有机分子占据而失去与臭氧分子的结合能力。
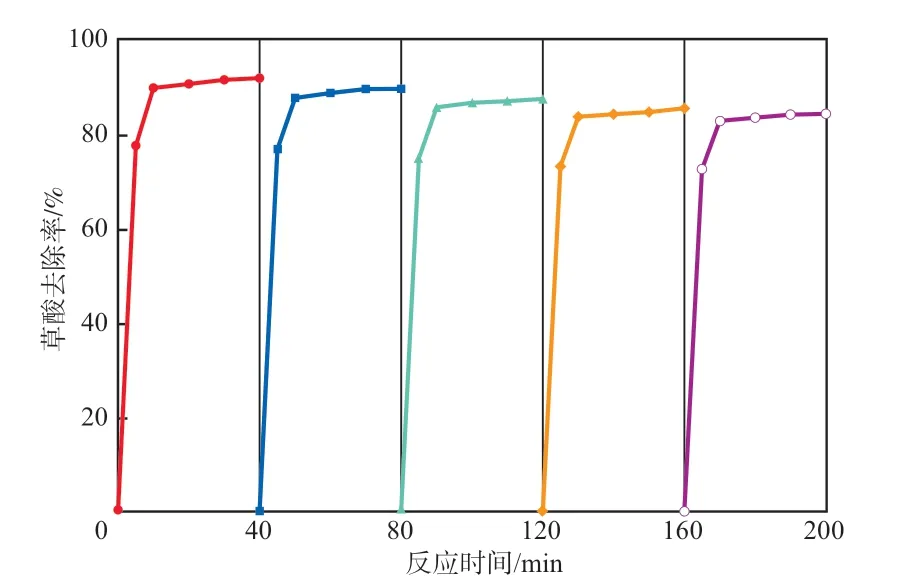
图8 催化剂的重复性实验
3 结论
a)采用水热—高温煅烧法制备了不同钴负载量的钴、氮、硼掺杂氧化石墨烯催化剂。其中,在钴负载量8%、焙烧温度500 ℃条件下制备的催化剂活性最高。在反应温度为323 K、pH为6.8、催化剂加入量为0.25 g/L、布洛芬质量浓度为50 mg/L、反应时间为10 min的条件下,布洛芬去除率可达99%。
b)该催化剂催化臭氧氧化降解布洛芬的过程符合准一级动力学模型,且催化臭氧体系的反应速率常数约为单独臭氧体系的5倍。
c)自由基探针实验结果表明,布洛芬的降解过程是一个自由基参与的氧化过程。矿化实验结果表明,该催化剂有利于促进污染物的深度降解。催化剂在重复使用5次后出现一定程度失活,但活性仍能达到初次使用的92.1%,表明该催化剂是一种活性强且较稳定的可再生催化剂。
