姐弟同患肝豆状核变性病例报道
2022-08-02蒋海燕赵悦郑荣秀
蒋海燕,赵悦,郑荣秀
(天津医科大学总医院儿科,天津 300052)
肝豆状核变性(hepatolenticulardegeneration,HLD)也称Wilson 病(Wilson′s disease,WD),是由13 号染色体上的ATP7B 基因突变导致细胞铜转运障碍的常染色体隐性遗传病,其特点是铜与铜蓝蛋白结合不足,胆汁排铜受损,导致铜在体内蓄积。该病主要累及肝脏、神经系统和精神系统,临床表现具有高度异质性,不同患者就诊时常表现出不同的症状,临床诊断较困难[1]。WD 在世界范围内的患病率约为1/50 000~1/30 000,各年龄段人群均可发病[2]。现结合相关文献对天津医科大学总医院收治的两例姐弟同患ATP7B 基因突变位点致肝豆状核变性的病例进行分析。
1 临床资料
病例1:患儿男,4 岁9 月龄,主因“确诊肝豆状核变性”于2021 年8 月就诊于天津医科大学总医院儿科门诊。入学体检发现肝功能异常,于外院行基因检测发现ATP7B 基因杂合突变,确诊肝豆状核变性。患儿为足月顺产,无窒息、产伤史,生长发育里程碑正常。父母均体健,奶奶为乙肝携带者。查体:身高111.6 cm(位于同年龄同性别同种族儿童第50~75 百分位);体重16 kg(位于同年龄同性别同种族儿童第10 百分位),体重指数(BMI)12.9 kg/m2。精神反应可,消瘦,心肺腹查体未见异常,体格发育无明显异常,肌力肌张力正常,神经系统查体未见异常。无精神异常相关表现。实验室及影像学检查:肝肾功能:谷丙转氨酶(ALT):324 U/L↑(5~40),谷草转氨酶(AST):273 U/L↑(8~40),碱性磷酸酶(ALKP):357 U/L↑(40~150),余项大致正常。乙肝两对半示保护性抗体;甲肝、丙肝(-),自身抗体:均为阴性;血氨:6.95 μmol/L↓(10~47),EB 病毒DNA、抗体:均未见明显异常;风疹病毒、弓形虫、巨细胞病毒、单纯疱疹病毒1+2 型IgM 均为阴性。腹部B 超:肝、胆、胰、脾、肾未见明显异常。24 h 尿铜:179.7 μg/24 h↑(15~60),染色体:46,XY,全外显子组测序检测发现 ATP7B 基因的 chr13:52511430c.4003G >C、chr13:52520505c.2975C>T,2 个杂合变异,分别来自父亲和母亲。予青霉胺100 mg tid[20 mg/(kg·d)]联合护肝治疗,低铜饮食。1 个月后复查:24 h 尿铜:611.4 μg/24 h↑较前排泄增多,血尿常规正常。肝肾功能ALT:339 U/L↑,AST:201 U/L↑,ALKP:339 U/L,较前好转,继予目前治疗。
病例2:患儿女,9 岁4 月龄,主因“发现肝功能异常”于2021 年8 月20 日就诊于天津医科大学总医院儿科门诊。患儿为足月顺产,无窒息、产伤史,生长发育里程碑正常。查体:身高140 cm(位于同年龄同性别同种族儿童第75~90 百分位),体重28.5 kg(位于同年龄同性别同种族儿童第50 百分位),BMI14.54 kg/m2。精神反应可,心肺腹查体未见异常,体格发育无明显异常,双乳腺Turner 2 期,肌力肌张力正常,神经系统查体均未见异常。无精神异常相关表现。实验室及影像学检查:血尿常规大致正常。肝肾功能:ALT:257 U/L↑,AST:113 U/L↑,余项大致正常。乙肝两对半:均阴性;24 h 尿铜:142.4 μg/24 h↑(15~60);铜蓝蛋白:4.37 mg/dL↓(22~58);血清铜:1.9 μmol/L↓(11~24)。颅脑MRI平扫:脑质未见确切异常;双侧上颌窦黏膜囊肿。腹部B 超:脾大、肝、胆、胰及双肾未见明显异常。裂隙灯检查未见Kayser-Fleischer(K-F)环。行基因检测发现ATP7B 基因chr13:52511430c.4003G>C、chr13:52520505c.2975C>T 存在2 个杂合突变,与其胞弟相同,遂确诊肝豆状核变性。予青霉胺190 mg tid[20 mg/(kg·d)]联合护肝治疗,低铜饮食。1 个月后随诊:血尿常规正常。24 h 尿铜:611.4 μg/24 h↑,较前排泄增加;肝肾功能:ALT:187 U/L↑,AST:90.9↑U/L,ALKP:230 U/L,较前好转。血β2 微球蛋白3.37 mg/L(1.3~2.7),胱抑素C1.63 mg/L(0.6~1.2),均稍增高。现治疗随访中。
病例基因检测结果见表1 及图1、2。
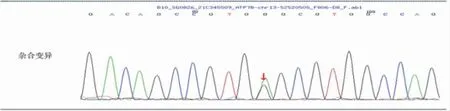
图1 ATP7B 基因chr13:52520505c.2975C>T

表1 基因检测结果
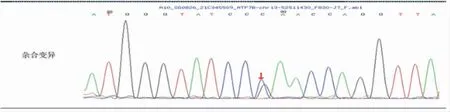
图2 ATP7B 基因chr13:52511430c.4003G>C
2 讨论
肝豆状核变性是由ATP7B 基因突变引起的,该基因位于染色体13q14.3,由21 个外显子和20 个内含子组成,该基因编码一个含有1 465 个氨基酸的铜转运p 型ATP 酶[3-4]。ATP7B 基因突变破坏ATP7B 铜转运蛋白的合成和功能,破坏铜的排泄途径,导致铜在体内异常沉积,主要累及肝脏、脑和角膜。肝豆状核变性多于青少年起病,以肝损害和神经系统损害为主要临床表现, 随访患者中最常见死因为肝衰竭[5]。肝脏是肝豆状核变性患者铜蓄积的初始部位,表现多种多样,包括无症状的生化异常和脂肪变性、急慢性肝炎、肝硬化以及肝衰竭。神经系统表现亦多种多样,包括构音障碍、肌张力障碍、震颤、帕金森综合征、舞蹈手足徐动症、脑性共济失调、认知损害、癫痫发作、各种精神和行为异常等。K-F 环是铜细小色素颗粒沉着于角膜后弹力层而形成的褐色环,反映了中枢神经系统中存在铜的沉积。其他器官铜沉积相关的异常包括:Fanconi 综合征等肾脏损害、早发性关节炎和软骨钙质沉着症特征的骨关节病、心肌病、肌病、胰腺炎、阳痿、不孕或反复自然流产及多种皮肤病等[1-2,6]。另有报道因“双下肢红色皮疹、瘙痒”等少见症状就诊的WD 患者,对于首发症状不典型者,临床医师要考虑到本病的可能性,避免患者因误诊造成的身体、心理及经济上的损害[7]。
既往WD 的诊断主要依靠临床表现和常规生化指标,包括24 h 尿铜升高、血清铜蓝蛋白低、肝铜含量升高。然而,生化检测可能具有误差。因此,分子遗传学检测对于建立WD 的精确和决定性诊断是有必要的,特别是在无症状患者和WD 患者的兄弟姐妹中[8-9]。全面的突变分析将提高对WD 遗传学的现有认知水平。特定突变与发病年龄和一些临床特征之间的相关性研究为基因型和表型之间的关系提供了新的见解。对大规模中国WD 队列中的基因型和表型相关性进行分析,进行DNA 提取和扩增,测序,发现突变率最高的外显子为8、13、16、12和18。这5 个外显子的总突变检出率为63.4%,表明这些外显子可能是重要的突变区域[10]。此两例患儿为外显子13,19 杂合突变,在肝豆状核变性患者ATP7B 基因突变的分布及频率图中显示Exon13 突变占17.3%,Exon19 突变占1%。肝豆状核变性的发病年龄及病情严重程度可能与基因突变的外显率、基因外因素以及环境因素的差异有关。本报道姐弟两人均以肝功能异常为首发表现,儿童期及青春期起病,没有行为异常和精神表现、神经系统症状和体征,腹部影像学检查未发现肝脂肪变性及结节状改变等肝硬化表现,头颅核磁均未见明显异常,可能与两人携带2 个突变位点均为杂合相关,也可能是发现较早疾病仍处于早期,治疗中需密切监测其他系统表现。同时密切观察药物的不良反应,定期检查血尿常规、肝肾功能、凝血功能、24 h 尿铜,肝脾B 超及颅脑MRI 等,以保证治疗效果及良好的预后。
肝豆状核变性是一种以全身铜积累为特点的遗传性疾病,随着现代医学的不断成熟,铜运输动力学以及肝铜沉积的新型标志物有助于明确诊断以及开发新的治疗方法。有研究采用正电子发射断层扫描(PET),以铜-64 为示踪剂,对WD 患者和对照组患者的肝铜代谢进行量化。结果显示在注射后20 h,WD 患者的标准化摄取值(SUV)高于对照组。显示铜PET 成像能够可视化和量化WD 患者的肝铜潴留的情况,为WD 病理生理学的研究提供了有价值的工具[11]。铜在肝脏中积累可导致肝脏脂肪变性甚至纤维化。体外研究表明,脂肪酸结合蛋白1(L-FABP)和脂滴相关蛋白5(PLIN5)与肝脏脂肪变性和纤维化有关。有学者研究WD 患者血清中LFABP 和PLIN5 水平与肝脂肪变性/纤维化的关系,结果显示,WD 组L-FABP 显著升高,L-FABP 浓度与肝纤维化有关。L-FABP 与转氨酶ALT 和AST 活性显著相关。血清L-FABP 可能是WD 儿童肝纤维化的一种新的生物标志物[12]。
WD 患者体内病理性铜积累引起毒性损伤、线粒体功能障碍和细胞凋亡,导致铜释放到血液中,从而影响其他器官。除去常见的肝脏、脑和角膜的沉积,铜沉积于心脏等重要器官亦引起关注。铜的积累对心肌细胞的毒性作用导致心肌功能障碍和结构重塑,以及对心肌组织和功能的影响及预后亦是研究热点之一。Grandis 等[13]在一项纵向队列研究中证实了WD 患者有较高的心力衰竭和心房颤动发生率,表明铜对心脏有潜在的不良影响。有研究探讨WD 患者心肌组织异常的发生率、表现和预测因素,应用心血管磁共振(CMR)分析心肌功能、应变和组织特征,对WD 患者进行分析,结果反映了在有神经系统受累症状或体征的患者中,患者发生心肌纤维化的风险较高[14]。总的来说,WD 的临床症状和疾病进展通常由肝脏和神经系统的表现驱动。因此,在WD 的早期阶段,除非患者出现了心脏病的症状或体征,否则可能不需要频繁进行心脏受累的特异性筛查。然而,随着疾病进展和神经系统受累症状或体征的发展,临床医师要关注心脏改变。
接受持续规范的排铜治疗或肝移植治疗并对治疗依从性良好的肝豆状核变性患者预后良好,尤其是在疾病早期,神经症状出现之前进行干预,即使是一些已发生晚期肝病的患者预后亦可[15-16]。除此之外还要对WD 患者的所有兄弟姐妹和子女进行筛查,及时的诊疗,准确寻找病因,减少各系统并发症的发生。最大限度的保证患儿的健康成长,仍是一项重大的临床挑战。
