杂多酸改性V-Mo/Ti-W催化剂的宽温SCR脱硝性能
2022-08-01周佳丽马子然赵俊平马静赵春林李歌王红妍王宝冬
周佳丽,马子然,赵俊平,马静,赵春林,李歌,王红妍,王宝冬
(1 北京低碳清洁能源研究院,北京 102211;2 国网能源和丰煤电有限公司,新疆 塔城 834411)
我国的能源结构以煤炭为主,煤炭燃烧向大气环境中排放大量的氮氧化物(NO)、二氧化硫(SO)、颗粒物(PM)和有害痕量元素汞(Hg)、砷(As)等污染物。其中NO(包括NO、NO和NO等)属于温室气体,是造成光化学烟雾、酸雨等环境污染的重要前体物,对大气污染影响严重,且危害人类生命健康。NO治理已受到政府和社会的广泛关注和重视,多种脱除NO的技术中氨气选择性催化还原(NH-SCR)已被证明是消除燃煤电厂烟气中NO最有前途和最有效的技术。
2014 年开始,我国煤电机组全面实施超低排放和节能改造工程,截至2019 年底,全国达到超低排放限值的煤电机组占全国煤电总装机容量的86%,NO排放量下降88.8%,煤电超低排放改造取得重大成就。“十三五”大气污染治理完美收官,2020 年,我国正式宣布力争2030 年前实现碳达峰,2060年前实现碳中和,全国大力发展风电、光伏发电等新能源。绿色发电的同时新能源大规模并网也带来了一系列挑战,由于风电、光伏发电具有随机性、间歇性和不稳定性特点,且电网负荷时差性较强,使得电网系统产生较大峰谷差,系统对灵活性电源需求不断提高,因此稳定电源火电机组将更多地承担系统深度调峰的角色。各地方政府也相继出台了针对火电机组调峰的补贴政策,激励火电机组参与灵活性调峰,实现全负荷运行。当燃煤电厂低负荷运行时脱硝入口烟气温度随之降低至250~300℃,目前市场广泛使用的钒钛基脱硝催化剂使用温度窗口仅为300~420℃,无法满足机组全负荷运行的NO超低排放要求。目前,解决脱硝系统全负荷运行的技术手段主要通过改造装置提高烟气温度和拓宽脱硝催化剂活性温度窗口两种途径。前者投资大,系统复杂,运行不够灵活且造成煤耗增加;后者通过使用宽温脱硝催化剂,具有投资运行成本低、运行灵活可靠等优点,逐渐受到人们青睐,在我国燃煤电厂的应用案例逐渐增多,但使用时最大的技术难点是需解决低温下烟气中生成硫酸氢铵而导致催化剂中毒失活的问题。
传统的脱硝催化剂以二氧化钛为载体,钒氧化物为活性组分,辅以三氧化钨为助催化剂的金属氧化物催化剂可在300~420℃温度区间内保持良好的脱硝活性,尽管可通过增加钒氧化物含量来提高催化剂在低于300℃下的脱硝活性,但随着VO含量的增加,VO物种在催化剂表面由多聚态逐渐团聚为结晶态物种,促使NH易被过度氧化形成NO、NO或NO,降低催化剂的N选择性,同时造成烟气中的SO向SO大量转化,SO易与NH反应生成硫酸氢氨(NHHSO),堵塞催化剂孔道,覆盖活性位点,造成催化剂活性降低,使用寿命缩短。因此,如何在不提高SO氧化率的同时提高低温下脱硝活性成为宽温催化剂研发的技术难点。不少学者通过引入金属(Nb、Sb、Nd、Ce、Se等)或类金属元素(B、Si、F等)来改善钒基催化剂低温脱硝活性,同时提高其抗硫性。张铁军等通过浸渍法制备Sb改性的VO-TiO催化剂,通过Sb 提高催化剂的氧化能力,增强催化剂的表面酸性,从而减缓催化剂表面硫酸铵盐的沉积,进一步提升催化剂的活性和抗硫性;李航航等采用溶胶-凝胶法和浸渍法制备了B 改性钒钛催化剂,结果显示,B改性可有效增大催化剂的比表面积和孔容,使催化剂表面弱酸量增加,进而表现出较高的活性和稳定性。可见对催化剂进行改性,增强表面酸性可明显提高钒基催化剂的催化活性和抗硫性能,但是已报道的改性催化剂对催化剂酸性的提高有限,因此其抵抗SO中毒的能力并不显著,其在高SO浓度下的脱硝性能尚未可知。
杂多酸(HPAs)具有独特的结构、超强酸性和不错的氧化还原特性,表现出良好的催化性能,因此被广泛应用于分析化学、表面化学、药物化学、电化学、食品化学、光化学和催化化学等领域。在所有杂多酸中,Keggin 型杂多酸(如HPWO、HSiWO和HPMoO)是最稳定且容易获得的,并且其笼形结构对极性分子具有强吸附性。因此,本文通过探索杂多酸作为常规V/Ti-W 催化剂的助剂,研究其拓宽催化剂温度窗口并提高抗硫性能的可行性,研究杂多酸和活性组分VO的协同作用,对设计制造工业可行的耐硫脱硝催化剂具有重要的工业化现实意义。为探索不同工业低成本杂多酸盐对V 基催化剂的性能影响,本文选用三种Keggin型杂多酸(磷钼酸、磷钨酸、硅钨酸),分别替代V-Mo/Ti-W催化剂中Mo和W的常规前体盐,制备了系列杂多酸改性的HPAs-V-Mo/Ti-W 催化剂,考察不同Keggin型杂多酸对催化剂的脱硝活性、N选择性、高硫工况抗硫性能测试及理化性能的影响。
1 实验
1.1 催化剂制备
采用等体积浸渍法制备V-Mo/Ti-W 催化剂。首先,将偏钒酸铵加入单乙醇胺溶液中搅拌溶解,待偏钒酸铵完全溶解后,将七钼酸铵加入溶液中,不断搅拌直至七钼酸铵完全溶解,然后将该溶液逐滴加入装有TiO-WO粉末的烧杯中,使其充分均匀地浸渍于TiO-WO粉(安徽迪诺环保新材料科技有限公司)孔道中至吸水达到恰好饱和,所得混合物在110℃下干燥12h,干燥后的样品置于马弗炉中400℃下高温煅烧5h,将煅烧后的催化剂进行研磨、压片、筛分,取40~60 目颗粒进行活性测试,样品命名为V-Mo/Ti-W。
对于杂多酸改性的V-Mo/Ti-W 催化剂,采用与上述相同的制备步骤,活性组分前体盐替换为杂多酸,以磷钼酸代替七钼酸铵的样品命名为HPMo-V/Ti-W;以磷钨酸或硅钨酸代替Ti-WO粉中W元素的添加,加入七钼酸铵、偏钒酸铵溶液中搅拌均匀至溶解后,逐滴加入装有TiO粉末(安徽迪诺环保新材料科技有限公司)的烧杯中,样品分别命名为HPW-V-Mo/Ti 和HSiW-V-Mo/Ti。所有催化剂样品中的V、W 和Mo 元素按VO、WO和MoO的质量分数计,含量相同。
1.2 催化剂表征
采用德国BRUKER AXS D8 型X 射线衍射仪进行X 射线衍射(XRD)测试,以CuK辐射为辐射源,扫描范围为2=10°~90°,扫描速度为8°/min;BET比表面积、孔容和孔径使用JW-BK222表面分析仪(北京精微高博)在77K 下使用N物理吸附测定,样品测试前在300℃真空下进行4h脱气预处理;H程序升温还原(H-TPR)和NH程序升温脱附(NH-TPD)表征测试在Auto ChemII 2920 吸附测试仪(美国Micromeritics)上进行;X 射线光电子能谱(XPS)使用具有1486.7eV Al K辐射的美国Thermo Scientific设备测量,并将284.8eV处的C 1s峰作为能量校正标准。采用德国布鲁克公司的傅里叶变换红外光谱仪对杂多酸改性催化剂进行分析。光谱仪分辨率为4cm,在波数为1200~700cm范围内进行扫描,KBr 压片制样,样品与KBr质量比为1∶100。
1.3 催化剂评价
催化剂脱除NO性能评价在固定床反应器(8mm)中进行,通过红外光谱仪(MKS,美国)在线监测反应进出口各气体浓度,每次测试催化剂用量为1mL 40~60 目颗粒,测试温度范围为200~375℃。模拟烟气组成为300μL/L NO、300μL/L NH、500~2000μL/L·SO、3% O(体积分数)、8%HO(体积分数),N作为平衡气,气体总流量为2500mL/min,体积空速(GHSV)为150000h。NO转化率和N选择性计算公式分别为式(1)和式(2)。
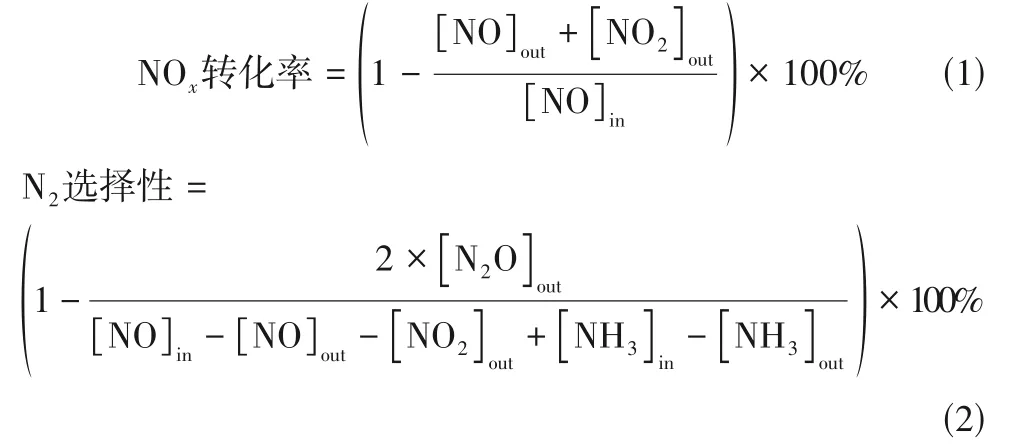
式中,[NO]、[NO]、[NO]、[NH]分别代表NO、NO、NO、NH反应器出口浓度;[NO]、[NH]分别代表NO、NH反应器进口浓度。
2 结果与讨论
2.1 NH3-SCR性能测试
图1(a)、(b)是改性催化剂以及V-Mo/Ti-W催化剂在不同温度下的脱硝催化效率和N选择性测试结果,每个温度点脱硝效率稳定后停留1h 再进行下一个温度点的测试。从图中可看出,在GHSV=150000h、同等活性组分含量条件下,经过杂多酸改性的催化剂在低温(200~300℃)下皆表现出比V-Mo/Ti-W 更高的NO转化率,提高幅度超过10%。其在250℃下的脱硝效率已超过70%,275℃超过80%,300~375℃超过90%,且在测试温度范围内始终保持接近100%的N选择性。相比之下,V-Mo/Ti-W 催化剂的N选择性在300℃以上逐渐下降,在375℃时下降到97%。因此,杂多酸改性不仅拓宽了SCR 催化活性的温度窗口,而且也显著减少了NO的产生,提高了N选择性,这对于减少温室气体NO对环境的破坏也有重要的促进作用。
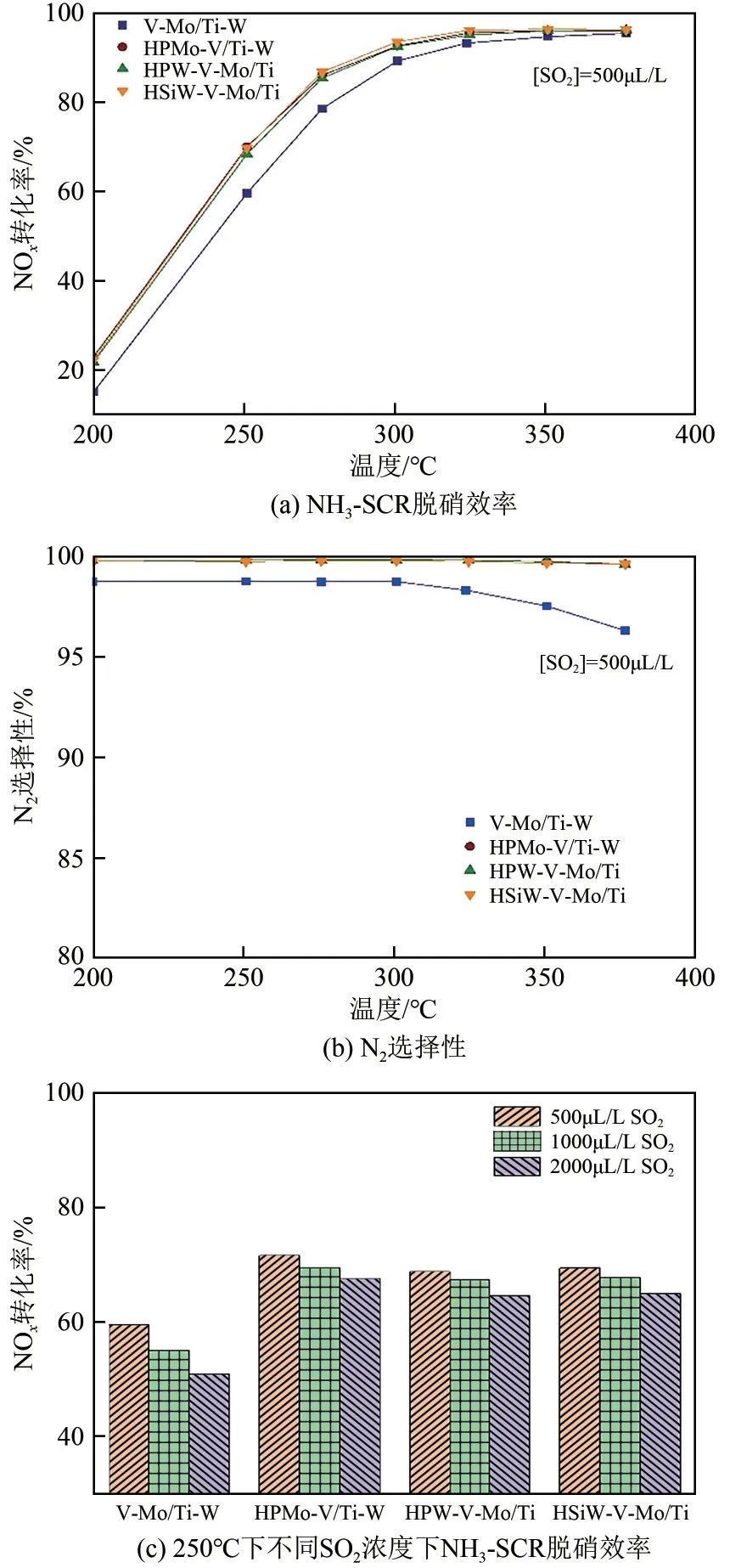
图1 杂多酸改性的催化剂及V-Mo/Ti-W催化剂的脱硝性能测试
燃煤电厂锅炉燃烧的煤种不同,烟气中的SO浓度(为200~2000μL/L)也不同,烟气中SO浓度越高,SO的产生量就越大,催化剂因生成的硫酸氢铵或硫酸氧钒的失效就越显著。如图1(c)所示,当SO浓度达到1500~2000μL/L 时,V-Mo/Ti-W 催化剂的脱硝效率急剧下降,对比之下,经过杂多酸改性的催化剂各温度下的脱硝效率随SO浓度的增加下降幅度较为轻微。这表明杂多酸改性的催化剂在燃煤电厂SCR 脱硝装置的宽温度范围内具有优异的抗SO性能。从催化活性的提高和抗SO衰减来看,磷钼酸的改性效果最好,磷钨酸、硅钨酸次之。
2.2 XRD性能测试
对杂多酸改性催化剂和V-Mo/Ti-W 做XRD 表征测试,以研究催化剂样品的晶体结构,结果如图2所示。分析发现,所有样品的衍射峰都归因于标准锐钛矿TiO(PDF-JCPDS-65-5724)的特征峰。未检测到五氧化二钒、三氧化钼、三氧化钨或磷钼酸、磷钨酸、硅钨酸的特征峰,说明钒和助催化剂皆以无定形状态存在或高度分散在载体表面上。与V-Mo/Ti-W催化剂相比,杂多酸改性的催化剂结晶度更低,这说明杂多酸与TiO载体之间存在较大的相互作用,从而抑制了其本身的团聚结晶,并降低了活性组分与载体的相互作用。文献报道,活性组分在催化剂表面高度分散有利于脱硝催化反应的进行。因此杂多酸改性后催化剂表现出更高的SCR 活性,与其促进活性组分VO的高度分散有关。

图2 杂多酸改性的催化剂及V-Mo/Ti-W的XRD谱图
2.3 FTIR分析
为确定杂多酸加入催化剂后保持以Keggin 结构形式存在,对杂多酸改性的催化剂在高温焙烧前后及脱硝反应稳定运行10h 后分别进行了FTIR 表征,结果如图3所示。据文献报道,Keggin型结构杂多酸的FTIR 特征吸收峰主要位于700~1100cm范围内,分别归属于(P—O)、(Si—O)、(M==O)、(M—O—M)、(M—O—M),M指Mo、W 金属原子,表示反对称伸缩振动频率。Keggin型结构的杂多酸各键反对称伸缩振动频率见表1。从图3 中可看出,通过等体积浸渍法制备的HPMo-V/Ti-W、HPW-V-Mo/Ti 和HSiW-V-Mo/Ti催化剂在高温焙烧及脱硝连续反应10h后,在890~850cm、940~960cm、1040~1100cm附近皆检测到归属于Keggin 结构的特征峰,说明经过高温焙烧,催化剂表面的杂多酸仍保持以Keggin 结构存在,但焙烧后的样品相较未焙烧样品的Keggin 结构峰稍向红移并强度减弱,推测是高温焙烧过程中,磷钨酸与催化剂表面其他含氧基团发生化学键合作用,以及杂多酸微量分解导致。对比杂多酸改性后的催化剂连续脱硝反应前后红外谱图,并无明显差异,说明Keggin结构在反应过程中较为稳定。
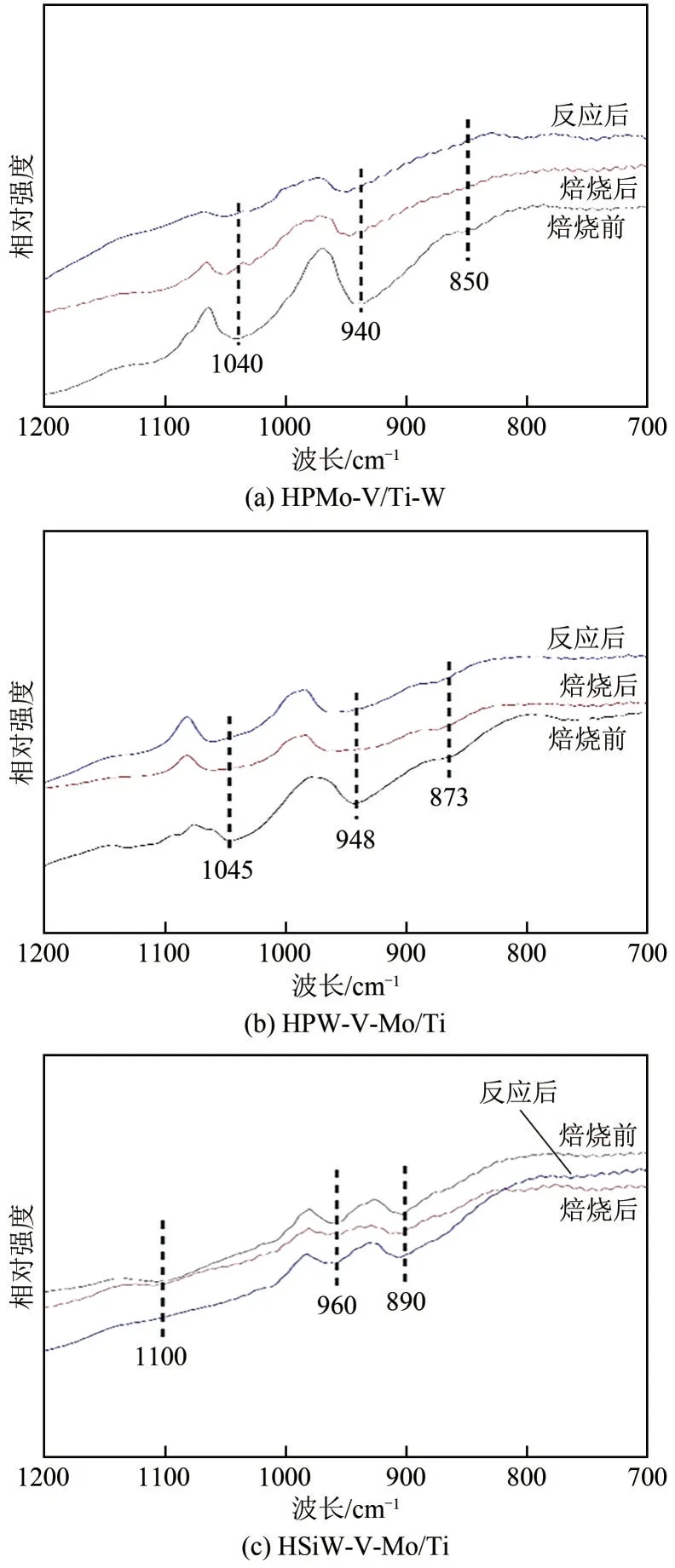
图3 杂多酸改性催化剂及V-Mo/Ti-W的FTIR谱图
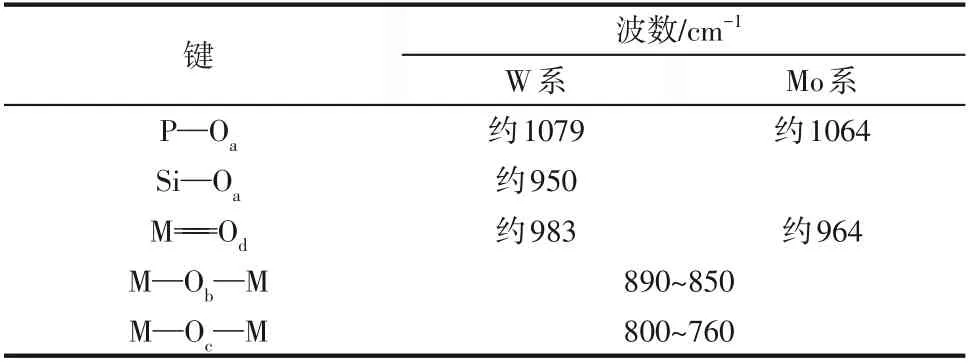
表1 Keggin型杂多酸各键反对称伸缩振动频率υas
2.4 BET比表面积及孔结构分析
催化剂样品及载体原料的BET 比表面积及孔结构参数列于表2,由于活性组分分散负载到载体表面且部分进入载体孔道中,催化剂样品的BET比表面积皆低于载体原料。对于催化剂样品,V-Mo/Ti-W、HPMo-V/Ti-W、HPW-V-Mo/Ti 和HSiW-VMo/Ti催化剂的比表面积分别为82.7m/g、83.2m/g、78.4m/g和78.9m/g,平均孔径分别为13.0nm、16.0nm、14.0nm 和14.2nm。总体来说,杂多酸改性对催化剂的比表面积影响不明显,HPMo改性后比表面积稍有增加,而HPW 和HSiW 改性后比表面积略有下降,但未改性的V-Mo/Ti-W催化活性相对最差,说明比表面积对催化剂活性影响较小。通常来说,较小的平均孔径可以使活性组分在催化剂表面分布更均匀,对催化反应有利。但因毛细管作用,当孔径较小时,硫酸氢铵在孔内的凝结变得更为容易。杂多酸改性后,促进了平均孔径的增加,有利于抑制ABS(硫酸氢铵)的凝结,从而预防催化剂硫中毒,这可能是由杂多酸特定的八面体笼形结构导致的。
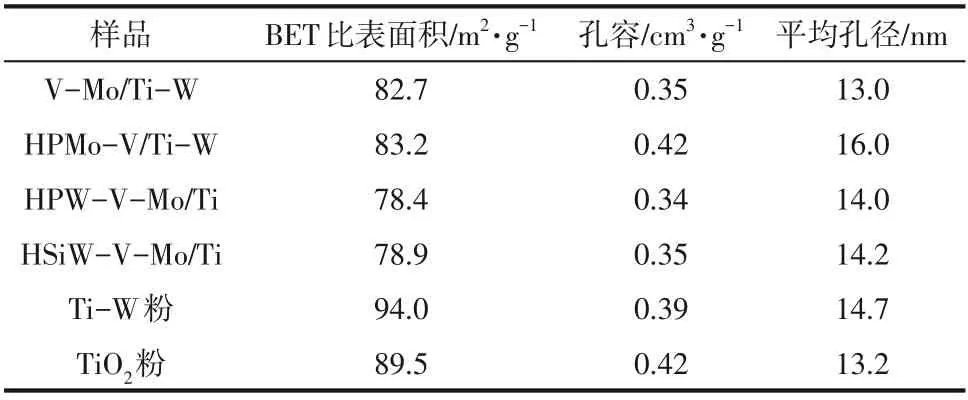
表2 杂多酸改性的催化剂及V-Mo/Ti-W的比表面积和孔结构
2.5 XPS分析
为了研究催化剂样品近表面区域中的O和V元素的化学组成和存在状态,通过XPS对其进行表征测试,结果如图4所示。对于O 1s图谱,通过峰值拟合,催化剂样品皆可分为两个峰,较低结合能(530.8~531.1eV)的峰值可归因于晶格氧(O,表示为O),而较高结合能(531.1~531.5eV)的峰值属于表面吸附氧(O或O,记作O)。低温脱硝反应中,催化剂表面吸附氧在催化反应过程中扮演重要角色,可促进NO 氧化为NO,此为该反应的关键速控步骤。而吸附态的NO和NO可与NH结合,形成不稳定的NHNO和NHNO物种,进一步迅速分解为N和HO,各催化剂表面吸附氧所占比例在表3中列出。从表3可见,杂多酸改性后的催化剂表面吸附氧比例相较V-Mo/Ti-W 催化剂明显提高,说明杂多酸改性可有效地增加催化剂氧空位含量,从而提高催化剂晶格氧到表面的迁移速率,增强NO的氧化活性,最终达到提高催化剂脱硝活性的目的。杂多酸改性样品中HPMo-V/Ti-W吸附氧占比最高,可能是其脱硝活性最高的原因之一。
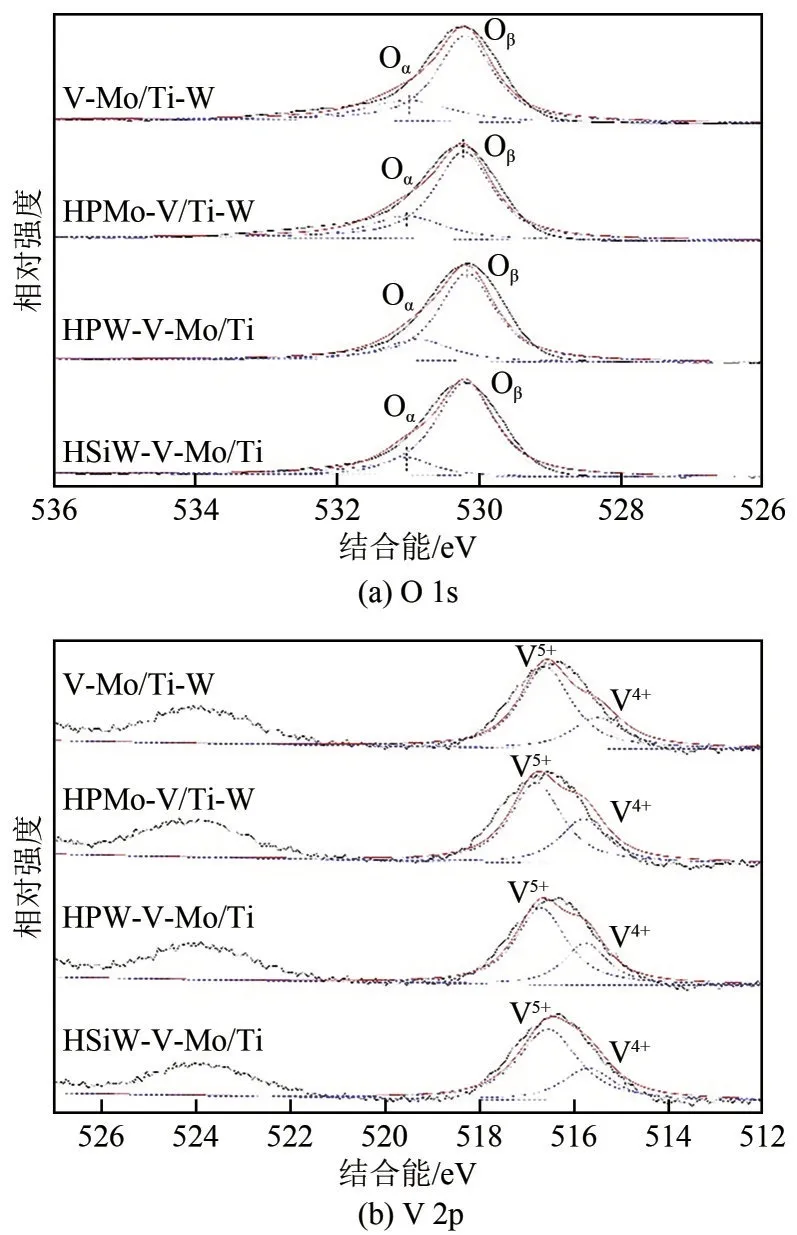
图4 杂多酸改性的催化剂及V-Mo/Ti-W的XPS谱图

表3 杂多酸改性的催化剂及V-Mo/Ti-W表面不同O和V元素原子比
图4(b)为不同催化剂V 2p轨道的XPS图谱。在电子结合能为516.7eV 和515.7eV 处的两个峰分别归属于V和V的特征峰。表3列出了不同催化剂表面V/(V+V)对比,结果显示,将杂多酸引入V-Mo/Ti-W 催化剂后,V物种占比提高。据文献[26]报道,在SCR 反应过程中,低价V 易吸附O生成活性氧物种,对提高催化活性有利。V 2p 的峰值位置向更高的结合能移动,这表明钒和杂多酸物种之间存在强相互作用,可以推断杂多酸改性的催化剂表面较高的V占比利于氧空位的产生,从而增加了表面吸附的O的量,这与上文分析的结果一致。
2.6 H2-TPR分析
为探究杂多酸改性对催化剂还原性能的影响,对催化剂样品进行H-TPR分析,结果如图5所示。V-Mo/Ti-W 催化剂在杂多酸改性前后皆显示三处明显的还原峰,分别集中在344~395℃、410~425℃和515~565℃范围内。低温处344~395℃和410~425℃的两个还原峰分别归属于载体上聚合态VO物种和孤立VO物种的还原。位于515~565℃之间的峰对应于W还原为W及Mo还原为Mo。与V-Mo/Ti-W 催化剂相比,杂多酸改性后的催化剂还原峰对应的还原温度皆向低温区偏移,说明杂多酸的加入提高了多聚态VO物种的含量,增强了催化剂的氧化还原能力,这与XPS 中V含量和O含量的增加相符。

图5 杂多酸改性的催化剂及V-Mo/Ti-W的H2-TPR谱图
2.7 NH3-TPD分析
催化剂的表面酸性对其催化活性有重要影响,本文通过NH-TPD 表征手段来分析杂多酸改性对V-Mo/Ti-W催化剂表面酸性的影响,结果如图6所示。杂多酸改性后的催化剂具有丰富的表面酸性,观察到低中高三个温度区域的脱附峰,而未改性的V-Mo/Ti-W 催化剂则只有低温和中温两个脱附峰。通常认为,低温(<200℃)下的弱酸性位主要归属于物理吸附的NH及热稳定性相对较差的B酸性位上的NH,中高温(>200℃)下的两个脱附峰则皆归属于较强L 酸性位中心结合的配位NH的脱附。所有样品的脱附峰峰温及对应的NH脱附量列于表4 中。NH脱附量与该温度区的酸量呈正相关,杂多酸改性后的催化剂NH脱附量皆高于VMo/Ti-W 催化剂,其中HPMo-V/Ti-W 样品最高,证明作为超强酸的杂多酸可显著增加催化剂的酸性位点,提高催化剂表面酸量。酸性的提高可以促进催化剂对NH的吸附能力,提高催化活性,并可抑制催化剂对SO的吸附,而提高催化剂的抗硫性能。这些因素与杂多酸改性后高硫工况下催化剂仍可保持高活性有关。

表4 杂多酸改性的催化剂及V-Mo/Ti-W不同温度下的NH3脱附量

图6 杂多酸改性的催化剂及V-Mo/Ti-W的NH3-TPD谱图
3 结论
采用等体积浸渍法制备了三种Keggin 型杂多酸改性的V-Mo/Ti-W 催化剂,通过FTIR 测试对比改性后催化剂在高温焙烧前后以及脱硝连续反应前后的Keggin 结构,结果显示高温焙烧后Keggin 结构有微量分解,而脱硝连续反应对Keggin 结构无影响。实验证明杂多酸改性可以显著提高V-Mo/Ti-W 催化剂在宽温度窗口(200~380℃)下,SO浓度为500~2000μL/L及8%HO含量工况中的NH-SCR 活性、抗硫性能和N选择性。通过各项催化剂表征对比发现,杂多酸对V-Mo/Ti-W 催化剂改性后,减弱了钒物种与载体之间的相互作用,产生更多的V和O物种,提高了催化剂的氧化还原性,促进了低温下NO的活化。杂多酸带来较多的酸性中心,提高了催化剂的酸量和酸强度,促使NH更稳定地吸附,不仅提高了全温段的脱硝活性,同时降低了高温下NO的产生。除此之外,杂多酸通过抑制SO的吸附,并通过其笼形结构抑制了硫酸氢铵的沉积,提高了催化剂在高SO烟气条件下的脱硝能力。该种催化剂具备优异的工业应用潜力,在我国煤电机组面临更多辅助电网消纳新能源并网发电的能源转型形势下,对保障脱硝系统宽负荷运行的灵活性具有重要意义。
