协同消除NOX 和二 英类物质催化剂的研究进展
2022-07-28余仕选牛晓巍魏政魏庆斌
余仕选 牛晓巍 魏政 魏庆斌
(沈阳大学区域污染环境生态修复教育部重点实验室,辽宁沈阳 110044)
1 引言
近几十年来NOX的消除成为社会关注的焦点。SCR 是目前最高效、稳定性最好的脱硝技术,SCR 系统被广泛应用于许多大型工业废气处理设施中,表现出优异的性能。然而,我国大气污染物的成分复杂,非电力行业排放大量的含氯挥发性有机化合物(CVOCs)和NOX,这些复合型污染物是形成PM2.5,O3及雾霾的主要前驱体,CVOCs 中通常涉及多氯同系物和二 英类物质(PCDD/Fs),对人类健康构成严重威胁。从宏观上看,近几年随着电力行业逐步实现超低排放,一些地区也陆续出台地方标准,进一步严格控制非电力行业污染物排放,如环境保护部等部门联合发布的《“十三五”挥发性有机物污染防治工作方案》指出,重点行业、重点地区VOCs 排放总量下降10%以上,通过与NOX等污染物的协同消除,实现环境空气质量持续改善。从行业需求上看,由于水泥、钢铁等非电力行业烟气中的CVOCs 浓度相当低,其浓度仅是NOX入口浓度的1/10,不可能单独建造一个装置来消除CVOCs。由于SCR 催化剂对CVOCs 有一定的去除能力[1],且NOX与CVOCs 消除的温度窗口重叠,因此科研人员尝试将SCR 技术进行升级改造后用于非电力行业多污染物的控制,不但节省成本,又节省空间。综上可知,无论是行业需求,还是国家政策导向,都使该领域催化剂的研发成为当前的热点。2018 年清华大学的研究者们在该领域开始调查研究[2-4],根据化学原理,利用过量的NH3和O2将NOX和CVOCs 转化成小分子的HCl,N2,H2O,CO2是可行的。
当前NOX和CVOCs 催化剂在实际运用中仍然面临着巨大的挑战,在多污染物协同消除的过程中,NOX的消除是一个还原过程,而CVOCs 的控制是一个氧化过程。多污染物控制催化剂的氧化能力太强可能使NH3发生过度氧化,导致NOX去除效率下降,而当多污染物控制催化剂的氧化能力太差时,CVOCs 不能完全氧化成HCl,COX(CO 和CO2)和H2O,从而会产生大量的多氯副产物,甚至还可能二次合成PCDD/Fs 等有害气体。因此对于催化剂活性组分的研发仍是当前多污染物控制催化剂的研究重点。本文综述了近几年工业烟气中NOX和CVOCs 协同消除催化剂的研究进展,并针对该研究提出一些建议。
2 PCDD/Fs 等氯代有机污染物的形成机理及控制技术
2.1 PCDD/Fs 等氯代有机污染物的形成机理
在工业生产中,了解PCDD/Fs 等氯代有机污染物的形成机理,有利于催化剂活性组分的优化,Dickson 等[5]使用五氯苯酚作为前驱体,用CuCl2作为催化剂,模拟PCDD/Fs 的形成,研究发现PCDD/Fs的形成量受加热时间、气体流速和温度的强烈影响。Deacon 反应提供了氯自由基,而氯自由基可能参与了PCDD/Fs 的形成,具体的反应方程式如下:
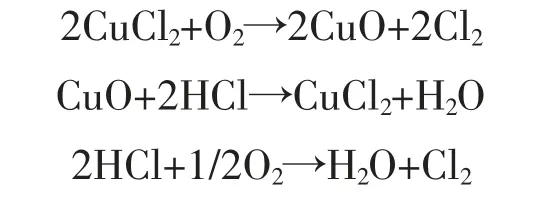
从以上方程式可知,铜元素在Deacon 反应中起着重要的催化剂作用,因此铜化合物对PCDD/Fs 的形成起着促进的作用。除了CuSO4催化活性低之外[6],CuCl2,CuO,CuBr2等铜化合物是工业热处理过程中形成PCDD/Fs 非常活跃的催化剂。因此在选择金属类催化剂时应该慎重选择含有铜源的化合物。PCDD/Fs 生成路线可以借鉴固废燃烧和热解过程中PCDD/Fs 的产生路线,在固废燃烧过程中目前公认的3 种PCDD/Fs 重新生成机理主要为[7-9]:
(1)高温气相反应。固体废物在燃烧的过程中会存在不完全分解的现象,分解后的烟气中还残留各种各样的碳氢类物质,这些物质与烟气中的含氯中间体或者氯的游离基通过聚合、缩合、环化等一系列化学反应后产生闭环的多氯化合物,例如多氯苯酚、多氯代苯等PCDD/Fs 的前驱体,这些前驱体在一些过渡金属催化剂(FeCl3,CuCl2)的作用下,在500~800 ℃的温度区间内非常容易进一步产生PCDF 或者PCDD。
(2)前驱物合成。固体废物在处置过程中,如果含有大量的PCDD/Fs 前驱物,例如氯苯、苯酚、环状的芳烃等化合物,这些小分子的化合物在金属离子(Cu,Fe)的催化作用下重组、缩合产生PCDD/Fs。
(3)从头合成。在燃烧后的低温区域内,大分子碳(残碳)与氧、氯、氢等元素通过基元反应,在合适的温度(200~400 ℃)和部分过渡金属离子催化作用下,经过一系列复杂的化学反应生成PCDD/Fs。
2.2 PCDD/Fs 等氯代有机污染物的控制技术
当前控制PCDD/Fs 产生主要有以下两项措施[7]:
(1)源头控制。工业生产原料中的一些苯系物、氯代物的前驱体及起作用的金属离子Cu2+,Fe3+等会影响多氯副产物的种类及浓度,因此在生产之前应该对这些原料进行筛选分类。
(2)催化还原法。催化还原技术被认为是目前最高效、运行最稳定的处理技术,主要用于烟气中NOX的脱除降解,在催化剂和NH3同时存在的条件下,其脱硝效率可高达90%。当前SCR 技术也被认为是降解含氯有机污染物最有希望的技术之一,针对SCR技术用于含氯有机污染物的研究,国外早已经进行了一些探索,1989 年德国Hagenmaier[10]首次发现SCR技术可将PCDD/Fs 催化降解成H2O,CO2,HCl 等无机的小分子化合物且不产生二次污染。一系列的实验表明,SCR 装置对烟气中排放的PCDD/Fs 有一定的处置能力,当PCDD/Fs 通过SCR 装置时,其总量和毒性当量能分别降低76%和69%。
3 NOX 和PCDD/Fs 多污染物控制催化剂的研究进展
3.1 MnCe 催化剂的研究进展
商用SCR 催化剂的工作温度通常在350~420 ℃之间,由于固体废物焚烧炉等工业烟气中实际排气温度低于300 ℃,因此商用的SCR 催化剂难以适应该温度窗口,低温催化剂的开发显得非常重要。当前Mn和Ce 的氧化物由于具有良好的储氧能力与氧化还原位点,氧化VOCs 时展现出较高的CO2选择性[11-12],当Mn 离子嵌入CeO2晶格中形成固溶体时,相比纯的CeOX和MnOX晶格氧的移动能力、比表面积和表面酸性都明显增强[12-13]。Gan 等[3]于2018 年采用浸渍法合成了不同比例的MnCe 催化剂,利用NO 和C6H5Cl(CB)模拟实际工业烟气中的NOX与PCDD/Fs多污染物,利用氯苯模拟实际工业烟气中PCDD/Fs是由于氯苯在降解的过程中会产生大量含氯副产物,其次PCDD/Fs 的毒性主要来源于C-Cl,且CB与PCDD/Fs 的相关系数达到0.93,因此氯苯常常被选为PCDD/Fs 的模拟分子[14-15],理论上多污染物催化剂只要对CB 去除有效,则也能同时去除PCDD/Fs等氯代有机物。
在NO 和CB 同时存在的反应气氛中,对不同比例的Mn ∶Ce 催化剂多污染物控制性能研究结果表明,当Mn∶Ce 为4 ∶6 时,对NO 和CB 协同消除效果最佳。在200 ℃时NO 和CB 的转化率分别达到约90%与25%,在275 ℃时NO 和CB 的转化率分别约为80%与90%,由此可见,MnCe 催化剂用于NOX和PCDD/Fs 协同消除时,NOX和CB 的活性区间不在同一个温度区域。
进一步研究CB 和NO 协同消除过程中的协同机制发现,当反应气氛中只通入NO 时,NO 在200 ℃的转化率基本达到100%,当温度大于200 ℃,NO 的转化率随温度的升高急剧下降。在275 ℃时NO 的转化率只有65%,相比多种气氛存在时下降了约20%,表明当温度高于200 ℃时,CB 对于NO 的SCR反应有一定的促进作用,这归因于NH3与CB 在催化剂表面发生了竞争性吸附,抑制NH3在MnCe 催化剂上发生过度氧化。而仅将CB 通入反应气氛中,在275 ℃时CB 的转化率为80%,当SCR 气氛存在时,CB 的转化率提升到90%,这归因于在该温度区间产生了一定量的NO2,NO2的氧化性比O2强,因此在多污染物控制反应中SCR 反应对CB 氧化起到促进作用。
MnCe 催化剂除了在高温阶段N2选择性差之外,还有一个显著的缺点是用于CVOCs 氧化时耐氯性较差,Cl 元素容易在催化剂表面沉积。Gan 等[16]用MnOX-CeO2同时控制NO 和CB 多污染物研究发现,从CB 上解离的Cl 容易通过亲电取代或亲核取代反应沉积在催化剂表面,导致催化剂的氧化还原性和SCR 表现降低。为了进一步研究Mn 基催化剂在NOX和CB 协同消除过程中失活的原因,Song 等[13]对MnCe 协同消除反应的稳定性进行测试,测试结果表明,MnCe 催化剂在低温条件下极易失活。通过对失活催化剂表面组分测试发现,催化剂表面发生了严重的积碳和积氯现象,其中碳沉积在催化剂的表面堵塞了催化剂的孔道,阻碍了反应物之间的接触,而氯沉积在MnCe 催化剂表面,主要是降低了Mn 元素的平均价态。进一步对比CB 和苯的氧化稳定性,测试结果表明,氯中毒是导致MnCe 催化剂失活的关键因素,而当SCR 组分存在时,MnCe 催化剂的失活现象有所缓解。
综上可知,尽管MnCe 催化剂在中低温条件下对多种污染物具有良好的转化率,但MnCe 催化剂在NOX和PCDD/Fs 协同消除反应中容易Cl 中毒失活,该缺陷是阻碍其工业化运用的关键。因此MnCe催化剂还需进一步优化,才有望实现在多污染物控制反应中的运用。
3.2 钒基催化剂的研究进展
3.2.1 钒基催化剂氧化还原性的调控
钒基催化剂可以同时去除NOX和CB,但在同一温度范围内无法实现对这两种污染物高效的去除。在低温(<300 °C)下,NOX还原速率较高,而CB 氧化反应速率较低。先前对MnCe 催化剂在多污染物控制中的反应研究表明,SCR 反应被促进是由于CB抑制NH3的过度氧化形成NO 或NO2,而CB 的氧化性能的提高是由于高温段NO2的产生,这些都与催化剂的还原能力相关,因此可以适当增加催化剂的氧化还原能力,缓解CB 对SCR 反应抑制的影响。Li等[17]设计了双活性位点PdV/TiO2催化剂用于NO和CB 的协同消除,经密度泛函理论(DFT)计算和实验表明,Pd 的掺杂促进了NOX,CB,NH3,O2的吸附,且Pd 引入后PdOX的形成和VOX与TiOX相互作用的增强导致氧的迁移速率加快,促进了协同控制中CB的氧化反应和SCR 反应。
WO3或MoO3常作为钒钛系催化剂的活性助剂,其不但使催化剂的活性和稳定性增强,同时还能提供额外的酸性位点[18-20]。Huang 等[21]于2018 年将WO3和MoO3分别添加到V2O5/TiO2,合成了一系列的VMo/Ti 和VW/Ti 催化剂。MPC 测试结果表明,VMo/Ti 催化剂相比于VW/Ti 催化剂展示出更高的SCR 反应和CB 氧化性能,在300~425 ℃能实现NO和CB 同时高效的去除,并保持良好的抗水抗硫性能。经DFT 计算和H2-TPR 测试表明,VMo/Ti 催化剂的能带间隙(1.16 eV)远低于VW/Ti 催化剂的能带间隙(1.49 eV),还原能力也优于VW/Ti,因此在MPC 反应中展现出更高的催化活性。Huang 等[21]相信V2O5/MoO3/TiO2可能成为NOX和PCDD/Fs 协同消除催化剂候选之一。
V2O5的含量会影响COX(CO 和CO2)、HCl 的选择性,同时当使用金属氧化物或者过渡金属氧化物进行CB 的氧化反应时,其PCDD/Fs 的二次生成是一个值得关注的问题。Huang 等[22]于2021 年分别制备了一系列VMo5/Ti,V3Mo5/Ti,V5Mo5/Ti,V7Mo5/Ti用于NO 和CB 多污染物控制影响机理的研究。在保持良好的抗水抗硫性能的前提下,V5Mo5/Ti 展现出最佳的SCR 反应和CB 氧化性能,其COX(CO 和CO2)、HCl 的选择性也与活性相一致,多污染物控制反应过程中形成的中间产物二氯马来酸酐(DCMA)随V2O5的含量增加而增加。在275 ℃条件下,使用GC/MS对V5Mo5/Ti 催化剂进行C6H5Cl 氧化反应尾气中17种具有毒性的2,3,7,8-TCDD 取代的PCDD/F 同系物生成情况进行了监测,结果表明,毒性最高的PCDD/F 同类物2,3,7,8-TCDD 没有被检测到,烟气中总PCDD/Fs 的浓度为0.011 5 ng I-TEQ/Nm-3,大大低于之前报道的VW/Ti 催化剂[23],因此在V5Mo5/Ti氧化CB 中产生的PCDD/Fs 可以忽略不计。
在多污染物控制过程中,钒基催化剂中VOX形态影响催化反应活性和中间产物的分布,当前被广泛接受的观点是,聚合VOX相比于单体VOX具有更高的还原能力,因此对NH3的活化能力更低,是低温状态下NH3-SCR 反应的决速步骤,而单体钒对于CVOCs 氧化反应更有利[24-25]。Zhai[1]课题组调查了单体VOX和聚合VOX协同消除NO 与CB 多污染控制过程中的反应特性,DFT 计算结合实验证实,CB在单体VOX上的吸附位点是V-OH,而聚合VOX上CB 的吸附位点是V=O,单体VOX为HCl 的解析提供充足的H+,而聚合VOX虽然有充足的氧空位,但催化剂表面容易形成V-Cl,容易发生亲电加氯现象。
3.2.2 钒基催化剂表面酸性位点的调控
催化剂表面额外的酸性位点能促进CB 的脱氯,而不是使多氯副产物在催化剂表面聚集。与VW/Ti 催化剂相比,V2O5/MoO3/TiO2(V2O5:5%,MoO3:5%)由于具有较好的多污染物控制表现和较低的PCDD/Fs二次生成量,可能成为最有潜力的多污染物控制催化剂候选之一,然而此催化剂的活性组分并不完美,在多污染物控制反应中大量的含碳或含氯副产物(CCl4和C2Cl4)积累在催化剂的表面,对催化剂的使用寿命有着巨大的影响,这暗示着虽然该催化剂具有良好的多污染物控制性能,但似乎含碳及含氯副产物在催化剂表面的积累不可避免。增强催化剂表面的Brønsted 酸和Lewis 酸不但可以增强NH3吸附的能力,额外的Brønsted 酸位点还能为HCl 提供H+,促进CB 的脱氯。而Lewis 酸的增加还可促进中间副产物的深度氧化,从而降低含碳和含氯副产物在催化剂表面的积累[17]。Marberger 等[26]通过瞬态实验与时间分辨实验,从分子水平上探究出钒基催化剂在SCR 反应中Lewis 酸是SCR 反应的活性中心,而Brønsted 酸中心没有参与循环反应。
本着该设计思路,Al2O3和SiO2作为常见的载体添加剂,不但可以增加催化剂表面的酸性、比表面积,还可以促进钒的分散[27-30]。Yu 等[31]用Al2O3和SiO2分别将V5Mo5/Ti 中TiO2的5%替换为Al2O3或SiO2,催化剂分别命名为Al-VMo/Ti 和Si-VMo/Ti。经多污染物控制反应测试,结果显示,Al2O3的添加同时提高了低温段(<275 ℃)和高温段(>400 ℃)的多污染物控制反应性能,经H2-TPR,XPS,NH3吸附原位红外、拉曼测试结果表明,低温段催化活性的提高可归因于催化剂还原能力的增加,高温段催化活性的提高可归因于Al2O3的添加促进了VOX的再分散,使Lewis 酸量明显增加,而SiO2的添加使其VOX的分散性变差,虽然其促进了Brønsted 酸量,但也导致Lewis 的损失,使其总酸量大大减少。通过TG-IR 实验分析催化剂表面残留的含碳和含氯化合物,结果表明,Al-VMo/Ti 催化剂表面没有发现有CCl4和C2Cl4生成,这对提高催化剂的使用寿命有着重要的意义。
4 问题与展望
当前针对NOX和PCDD/Fs 多污染物控制催化剂的研发还处于起步阶段,多污染物协同消除反应中各气体组分之间的影响机理以及多污染物控制反应过程中副产物的分析虽然取得了一定的研究进展,但是针对NOX和NH3与CB 氧化后的中间体有机物碎片反应的机理尚不明晰,其次是从CB 中脱下的Cl 在反应中具体的迁移路径尚不明晰,可通过DFT 计算与实验结合进一步阐述其反应机理及路径。
前面提及的Al-VMo/Ti 虽然有良好的SCR 反应性能及大量的CB 脱氯位点,但是该催化剂在多污染物控制反应中CO 的选择性仍然占COX选择性的50%左右,因此V5Mo5/Ti 催化剂在多污染物控制反应的性能还有提升的空间。由于贵金属性质活泼,对于VOCs 具有良好的氧化能力。Liu 等[32]研究了CB在贵金属催化剂(Pd/TiO2,Pt/TiO2,Ru/TiO2,Rh/TiO2)氧化多氯副产物的分布情况,结果表明,贵金属Ru/TiO2催化剂对CB 具有较高的氧化能力,同时CO2的选择性也比较高,相比于其他贵金属催化剂,Ru/TiO2形成的多氯副产物最少。经XPS 分析表明,Ru/TiO2表面残留的吸附Cl 和有机Cl 总和的比值最小,形成的PCDD/Fs 也是最少,因此这意味着贵金属Ru 具有良好的Cl 元素移除能力,表明不会发生Cl 中毒现象。因此在未来NOX和PCDD/Fs 多污染物协同消除催化剂的开发中,可以尝试适当引入贵金属Ru,既可以提高CO2的选择性,又能促进多氯副产物的深度氧化。
综上可知,当前NOX和PCDD/Fs 多污染物协同消除催化剂是非电力行业研究的一个热点,有着广阔的发展空间。
