加权基因共表达网络分析慢性鼻窦炎伴鼻息肉发病机制
2022-07-27刘萌芷叶林峰
陈 哲 肖 伟 刘萌芷 彭 涛 叶林峰
武汉大学中南医院耳鼻咽喉-头颈外科 湖北 武汉 430071
加权基因共表达网络分析(weighted gene co-expression network analysis,WGCNA)是一种分析多个样本的基因表达模式的方法。主要采用将表达模式类似的基因进行聚类,形成一系列模块(module),随后将模块与特定性状或表型间的关联进行进一步分析得到有生物学意义的数据。该分析方法通过寻找协同表达的基因模块,计算基因网络与关注的表型之间的相关度,发现网络中的与其他基因连接度最高且与相应的临床特征关系最为密切的核心基因(Hub genes)。本研究拟结合WGCNA 方法计算并筛选出慢性鼻窦炎伴鼻息肉发病过程中的关键模块,挖掘核心基因。对核心基因进行基因本体分类(gene ontology,GO)分析和京都基因与基因组百科全书(Kyoto Encyclopedia of Genes and Genomes,KEGG)通路富集分析,了解其分子功能和生物学过程。
1 资料与方法
1.1 数据收集从美国国立生物信息技术中心(National Center for Biotechnology Information,NCBI)的基因表达综合数据库(Gene Expression Omnibus,GEO)网站下载表达谱数据GSE36830,数据集在Affymetrix 人类基因组U133 Plus 2.0 阵列平台上执行。其中包含24 个样本,正常对照组钩突,慢性鼻窦炎不伴息肉组钩突,慢性鼻窦炎伴息肉组钩突及息肉各6 例样本。
1.2 构建加权基因共表达网络主要采用R 包“WGCNA”,依据使用要求并结合GSE36830 样本基因表达及样本分组相关数据进行分析。
1.2.1查看样本数据的完整性,剔除离群值 为了避免离群值对后续计算的影响,在构建加权共表达网络前需保证样本数据的一致性,当样本中存在显著的离群值时将其剔除。采用WGCNA 包自带函数验证样本的完整性,剔除存在过多缺失值和显著脱离其他样本的离群样本。
1.2.2确定软阈值、构建共表达矩阵及模块识别WGCNA 算法采用相关系数加权值,使得网络中的基因之间的连接服从无尺度网络分布(scale-free networks)。利用“WGCNA”包中pickSoftThreshold函数来进行软阈值(soft threshold power)的确定,本研究中将相关系数R2>0.9 作为确定软阈值β的标准。采用一步法构建共表达矩阵,得到表达模式相似的模块,并将其可视化。
1.2.3识别与临床分组相关的模块,核心基因筛选 分别计算不同模块与各临床分组间的相关性,从中筛选与相关疾病相关性高的模块。计算被选中的模块中的基因与临床疾病的相关性,并依据模块身份(module membership,MM)与基因显著性(gene significance,GS)进一步筛选核心基因。以|MM|>0.8 且|GS|>0.2 作为标准筛选核心基因。
1.3 核心基因GO 分析对进一步筛选出的核心基因进行功能注释,即GO 分析。分别从生物学过程(biological process,BP)、细胞成分(cellular component,CC)及分子功能(molecular function,MF)三个层面来分析核心基因的作用。本研究中采用R软件中的“cluster Profiler”包对筛选的所有核心基因进行GO 富集分析。
1.4 核心基因KEGG 分析KEGG 是从分子水平,特别是以基因组测序以及其他高通量测序技术的结果建立的数据库资源。KEGG 通路分析对分子间相互作用的网络通路有较好的呈现。对上述筛选的模块中的核心基因,利用R 软件中的“Cluster Profiler”包进行KEGG 通路分析。
1.5 核心基因差异表达分析对筛选出的核心基因的表达量进行差异分析,采用R 软件中limma 包筛选核心基因中差异表达的基因[1]。比较慢性鼻窦炎伴鼻息肉中息肉组织与其他各组钩突组织的基因表达量。其中以表达量倍数(fold change,FC)为上 调 或 下 调 2 倍 即 |log2FC| ≥1,调 整 后P(adj.P)<0.05 作为存在差异表达的核心基因。
2 结果
2.1 构建无尺度网络及模块分类计算任意两个基因间的相关系数,设置阈值进行筛选,使得基因之间的连接符合无尺度网络分布。随后将基因按表达模式进行模块分类。以相关系数R2>0.9 为标准,选择合适的软阈值,本研究中的软阈值β=3(图1A、1B),并依据此值建立临近矩阵及拓扑矩阵。当β=3 时,计算每个基因的连接度。在无尺度网络中的连接度存在幂律分布,本研究中k 与p(k)之间存在负相关,因此构建的网络符合无尺度网络分布(图1C、1D)。构建共表达矩阵后,可以发现所有基因依据其表达类型的相似性被归为不同的模块,共27 个模块(图1E)。
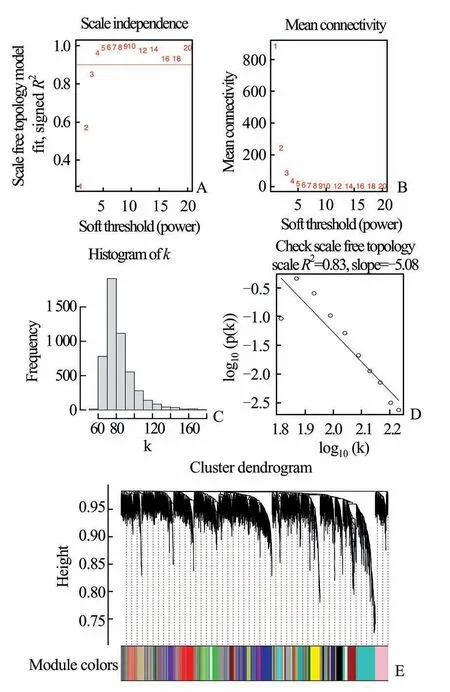
图1 构建无尺度网络及模块分类
2.2 慢性鼻窦炎伴鼻息肉相关模块的筛选依据模块的特征向量,计算模块与慢性鼻窦炎伴鼻息肉的关系,与之相关性最大的模块为棕色模块,共包含392 个基因,相关性为0.68(P<0.01)(图2A)。同时将不同模块基因特性与性状进行聚类分析后,并进行热图可视化,也可发现慢性鼻窦炎伴鼻息肉与棕色模块高度相关(图2B)。随后分别对所有模块包含的基因与该模块进行相关性分析,其中棕色的相关性仍为最高(图2C)。以|MM|>0.8 及|GS|>0.2 作为棕色模块中核心基因的筛选条件,共筛选出24 个基因(图2D,右上象限)。
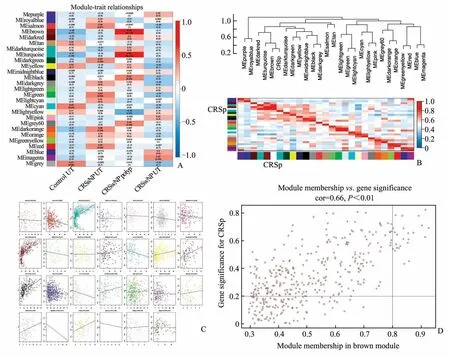
图2 慢性鼻窦炎伴鼻息肉相关模块的筛选
2.3 核心基因GO 分析对筛选出的核心基因进行GO 分析。其中BP 与细胞免疫关系密切,CC 显示核心基因主要作用于细胞膜,MF 分析可看出核心基因与细胞因子、趋化因子的活性及相关受体等密切相关(图3)。
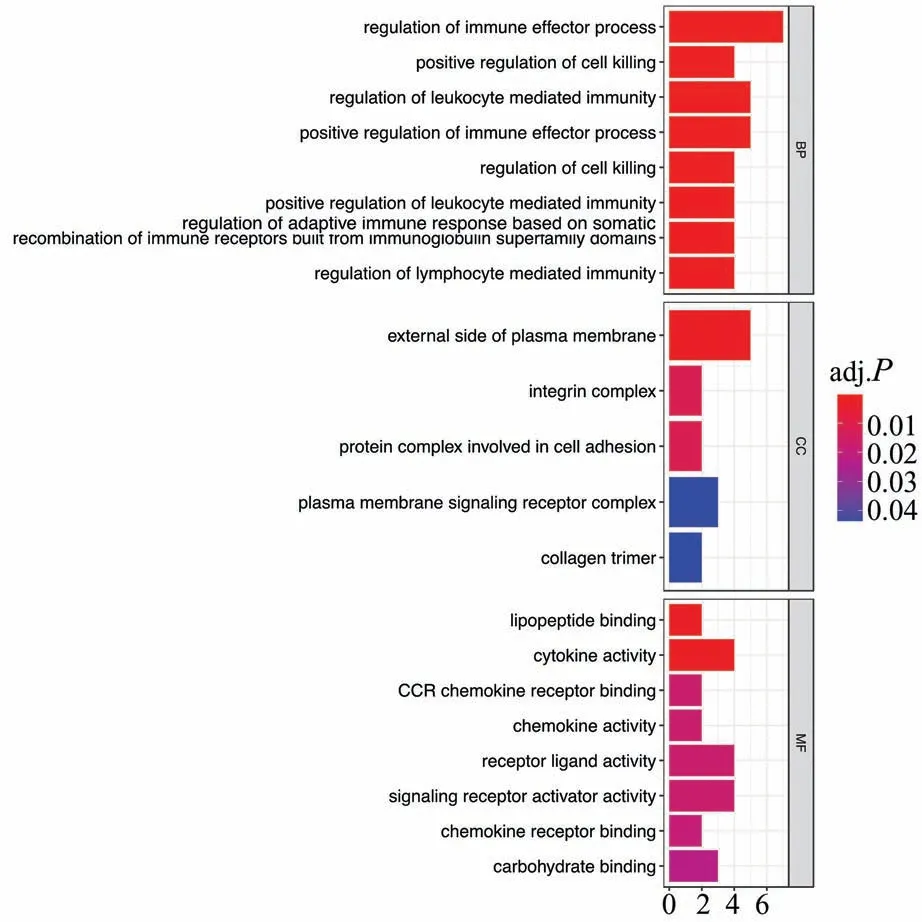
图3 GO 分析结果
2.4 核心基因KEGG 分析从KEGG 分析结果来看,核心基因主要作用于造血细胞谱系、病毒蛋白与细胞因子和细胞因子受体的相互作用、阿米巴病、细胞因子与细胞因子受体的相互作用、Th1 和Th2 细胞分化、紧密连接、JAK-STAT 信号通路、趋化因子信号传导途径等(图4)。
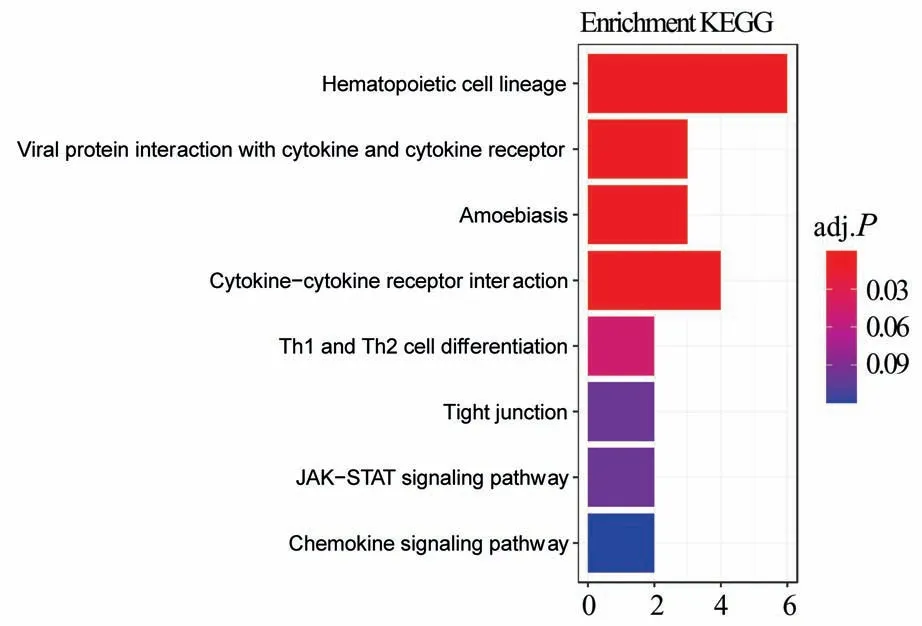
图4 KEGG 分析结果
2.5 核心基因的表达量差异分析通过R 软件“limma”包对核心基因进行表达量差异分析后,以表达量差异倍数为2,调整后P<0.05(|log2FC|≥1,adj.P<0.05),共筛选出15 个差异表达的基因(CCL22、GFI1B、ITGAM、FCER2、CLEC4GP1、CD1E、ALPK2、VSTM1、AOC1、MGARP、IL2RA、COL6A5、CD1A、CLC、SIGLEC8)(图5)。
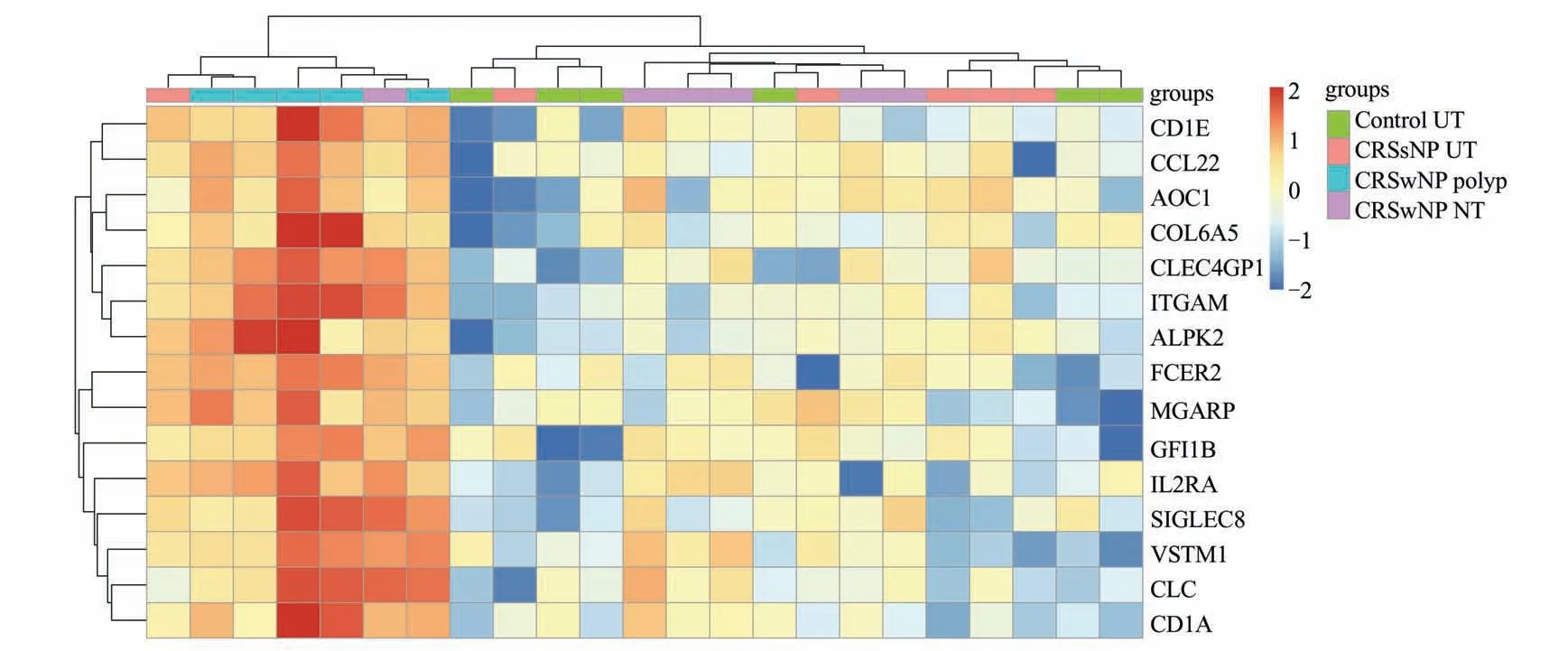
图5 棕色模块核心基因中差异表达基因的表达量热图
3 讨论
慢性鼻窦炎(chronic rhinosinusitis,CRS)是耳鼻咽喉头颈外科临床常见疾病之一。流行病学调查显示我国慢性鼻窦炎总体患病率约为8.0%,影响了约1.07 亿人,带来了严重的经济及社会负担[2]。慢性鼻窦炎伴鼻息肉(chronic rhinosinusitis with nasal polyps,CRSwNP)是慢性鼻窦炎的一种常见类型,患者临床症状一般较重,相较于不伴鼻息肉的慢性鼻窦炎患者,术后复发相对较高。由此可以推断慢性鼻窦炎不同的临床分型其内在的分子生物学机制可能存在差异。针对不同疾病类型的患者进行个体化的医疗过程的精准医疗将会成为疾病治疗的趋势。精准医学的发展离不开基因测序技术快速进步以及生物信息与大数据科学的交叉应用。生物信息学技术可以从数量巨大的测序数据中对有效数据进行整合及分析,计算出可能在生理和病理过程中发挥重要生物学功能的数据。
采用生物信息学方法挖掘慢性鼻窦炎伴鼻息肉的关键基因能为疾病的发病机制的研究提供新的思路。WGCNA 算法被应用在多种疾病中应用广泛,如肺癌[3]、乳腺癌[4]及变应性鼻炎[5]等。本文通过WGCNA 方法分析出在慢性鼻窦炎伴鼻息肉中存在显著生物学意义的基因,筛选出在该疾病表型中表达模式类似的基因模块,并通过进一步的筛选,找出模块中的核心基因。通过对核心基因进行GO 分析,可以看出核心基因主要作用于细胞膜,与细胞免疫关系密切,通过调节细胞因子、趋化因子的活性及相关受体等发挥生物学作用。KEGG 分析则可以预测慢性鼻窦炎伴鼻息肉的发生可能与细胞因子与细胞因子受体的相互作用、Th1 和Th2细胞分化、紧密连接、JAK-STAT 信号通路、趋化因子信号传导途径密切相关。
对核心基因进行更进一步表达量分析,发现15个在慢性鼻窦炎伴鼻息肉组与其他组间存在显著表达差异的基因。其中CCL22、ITGAM、CD1A、CLC、SIGLEC8 参与了慢性鼻窦炎的发病过程[6-9]。而尚未发现GFI1B、FCER2、CLEC4GP1、CD1E、ALPK2、 VSTM1、 AOC1、 MGARP、 IL2RA、COL6A5 在该疾病中的相关研究。进一步的文献研究发现GFI1B 是造血系统的关键调节因子[10];FCER2 编码IgE 受体亚单位,可影响血清总IgE 水平[11];ALPK2 与心肌细胞及某些肿瘤关系密切[12];VSTM1 与白血病等有关[13];IL2RA 作用于T 细胞活化过程[14];MGARP 在类固醇生成组织和视觉系统中高度丰富;AOC1(ABP1)与组胺的产生及降解等 相 关[15];COL6A5 与 特 应 性 皮 炎 关 系 密 切[16]。CLEC4GP1 目前研究较少,可能与胶质母细胞瘤相关[17]。从相关基因在其他疾病中的研究可以推测大部分基因与造血细胞及免疫系统密切相关,与KEGG 预测的结果有较好的一致性。
综上,可以推测加权基因共表达网络分析可以通过相关计算,挖掘出在慢性鼻窦炎伴鼻息肉发病机制中可能存在重要生物学意义的新基因,并可以初步推断出相关基因可能的作用机制,并从其他学者的研究中证实了相关基因的功能。在后续的工作中需进一步对上述未在慢性鼻窦炎伴鼻息肉中得到充分研究的相关基因进行探索。
