马方综合征的最新诊疗进展
2022-07-25杨书婷综述罗芳审校
杨书婷 综述 罗芳 审校
(浙江大学医学院附属第一医院儿科,浙江杭州 311100)
马方综合征(Marfan syndrome,MFS)是一种常染色体显性遗传的多系统结缔组织疾病,常累及心血管、骨骼和眼部,也可累及中枢神经系统、呼吸系统和皮肤组织等,90%~95%的MFS 由纤维蛋白原‐1(fibrillin‐1,FBN1)基因突变所致,少数由TGFBR1基因或TGFBR2基因突变引起[1-2]。
FBN1基因位于15q21.1,由65个外显子组成,编码的FBN1蛋白分子量约320 kD,是一种弹性蛋白相关糖蛋白,是细胞外基质中微纤维的主要成分,维持着弹性纤维的长期稳定性,在心血管、骨骼、眼等组织器官中发挥重要作用,截至2020年3 月,已报道3 000 多名不同的MFS 患者,已发现FBN1基因1 800 多种致病性变异,以错义突变常见,其次为无义突变、微小插入或缺失突变,拷贝数变异和其他结构变异也可导致该病[3-4]。FBN1基因有3个主要结构域,包括47个表皮生长因子样结构域、27个潜在转化生长因子结合蛋白结构域和微纤维蛋白结构域,这些结构域在防止FBN1蛋白水解、促进FBN1单体与其他微纤维组分之间的相互作用及稳定微纤维结构方面发挥重要作用,微纤维通过弹性层与平滑肌细胞和内皮下基底膜的相互连接维持主动脉的结构稳定。FBN1 蛋白可通过潜在转化生长因子结合蛋白调节转化生长因子β(transforming growth factor β,TGF‐β)的生物利用度,当FBN1基因突变,不能编码FBN1蛋白,TGF‐β的信号转导被干扰,蛋白降解、弹性纤维结构与功能破坏,导致广泛的血管重塑、结缔组织的进行性破坏,以及主动脉瘤及心脏瓣膜病变的发生[5-6]。
儿童与成人型MFS 发病率约1/5 000,25%~35%为基因新发突变,在积极正确地管理下,其预期寿命可与普通人群接近。新生儿马方综合征(neonatal Marfan syndrome,nMFS)的发病率约1/10 000,远低于儿童和成人型MFS,92%为基因新发突变,临床表型重,预后差,多于婴儿期死亡[7]。本文就MFS 目前的诊疗进展作一综述,期待能进一步提高临床医护对MFS的认识,早发现、早干预,尽可能使MFS患儿获得良好预后。
1 MFS的诊断
1.1 临床特征
MFS 发病无种族与性别差异,由于基因组印记、表观遗传修饰、环境影响等原因,患者临床表型多样,主要表现为主动脉根部进行性扩张、晶状体异位、骨骼畸形,还有主动脉其他部位扩张、动脉瘤、心脏瓣膜脱垂、拇指征及手腕征、骨质疏松、脊柱侧凸、肌病、漏斗胸、近视、视网膜脱离、青光眼、硬脑膜扩张、自发性气胸、皮肤褶皱等表现[8-10]。2019年Laganà等[11]使用3D几何模型测量分析了MFS 受试者的腭形态,发现MFS 患者存在高腭弓、上颌收缩等颅面畸形表现,与MFS 患者在睡眠期间上气道塌陷、阻塞性通气障碍有关,其上颌生长模式尚需进一步研究。儿童MFS 以主动脉窦部扩张常见,可合并二尖瓣脱垂,与成人期严重的二尖瓣关闭不全和心内膜炎发生有关[12-13]。nMFS 极为罕见,除与经典型MFS的共有特征外,还有肢体屈曲挛缩、“老年”貌等特征,多数在产前检查时已能发现相关异常[14]。经典型MFS的主要死亡原因为主动脉夹层或破裂,而nMFS的主要死亡原因是二尖瓣和/或三尖瓣关闭不全及主动脉根部扩张相关的充血性心力衰竭、肺动脉高压。
1.2 辅助检查及基因检测
超声心动图、心脏CT、心脏MRI 是诊断MFS的检查手段。心脏CT 血管造影能确定动脉瘤的扩张程度,但存在一定局限性,如在升主动脉中可能存在心脏运动产生的伪影,且有辐射性,不适合多次检查。心脏MRI 无辐射性,且较CT 有更好的检查效果。
MFS 为常染色体显性遗传,基因检测有助于诊断及鉴别诊断,常用的测序方法包括Sanger 测序、二代测序、多重连接探针扩增技术[2,15-16]。在nMFS 中,约86%的FBN1基因突变位点聚集在外显子24~33 内,该区也被称为“新生儿区”,研究者发现位于24~33号外显子突变热点的先证者,病死率高,且具有严重的心血管系统表型,预后较差[7]。但2020 年Willis 等[17]报道了1 例24岁存在FBN1G1013R 突变的MFS 男性患者,突变位点位于25 号外显子,其9 月龄时发现二尖瓣脱垂、主动脉根部扩张,但无临床表现;18 月龄时因反复呕吐出现进行性呼吸窘迫及室上性心动过速,心脏彩超示腱索断裂;3岁行二尖瓣置换术,8岁行主动脉根部置换术,后未再行手术治疗;目前仅口服药物,仅有轻度运动不耐受,不影响生活质量,该报道指出FBN1G1013R 突变发生在高度保守区域,且已在其他4名无遗传关系的MFS患者中发现相同突变,其出现心脏受累的时间均较晚,预期寿命均较长,因此,研究者提出1岁前有MFS表现和早期心脏受累的婴儿,其预后将非常差,携带FBN1G1013R 突变的患者可能预后良好,但不能排除手术干预产生的影响,也不能排除是否有其他基因保护机制降低了此类患者的病情严重性。2021 年Arnaud 等[4]评估了1 575 名MFS 患者及其亲属之间基因型和表型的相关性,发现提前终止密码子突变患者的主动脉事件及严重脊柱侧弯的发生风险较高,预期寿命缩短,但晶状体异位的发生风险较低;另外,半胱氨酸参与二硫键的形成,对维持FBN1蛋白三级结构的稳定性具有重要作用,根据框内突变对FBN1蛋白半胱氨酸含量的影响,可初步评估疾病严重程度。Stengl等[5]提出了可预测主动脉受累程度的指标,分别为血液循环中TGF‐β 水平、半胱氨酸水平、动脉迂曲度、主动脉生物力学参数、基因型-表型相关性等,但还需大量前瞻性的研究支持,并且由于MFS 致病基因突变的多样性及临床表现的异质性,目前MFS的基因型及临床表型的关系尚无定论[18]。
1.3 诊断标准
目前国际上关于MFS 的诊断标准是根特标准(Ghent criteria),1996年首次定义,2010年重新修订[19-20](表1),更新后的根特标准强调主动脉根部扩张、晶状体异位的重要性,由于某些特征的外显率与年龄有关,在儿童患者中需谨慎使用根特标准[7]。系统评分(表2)用于评估MFS患者的特征性症状,总分20分,7分以上有诊断价值[20]。
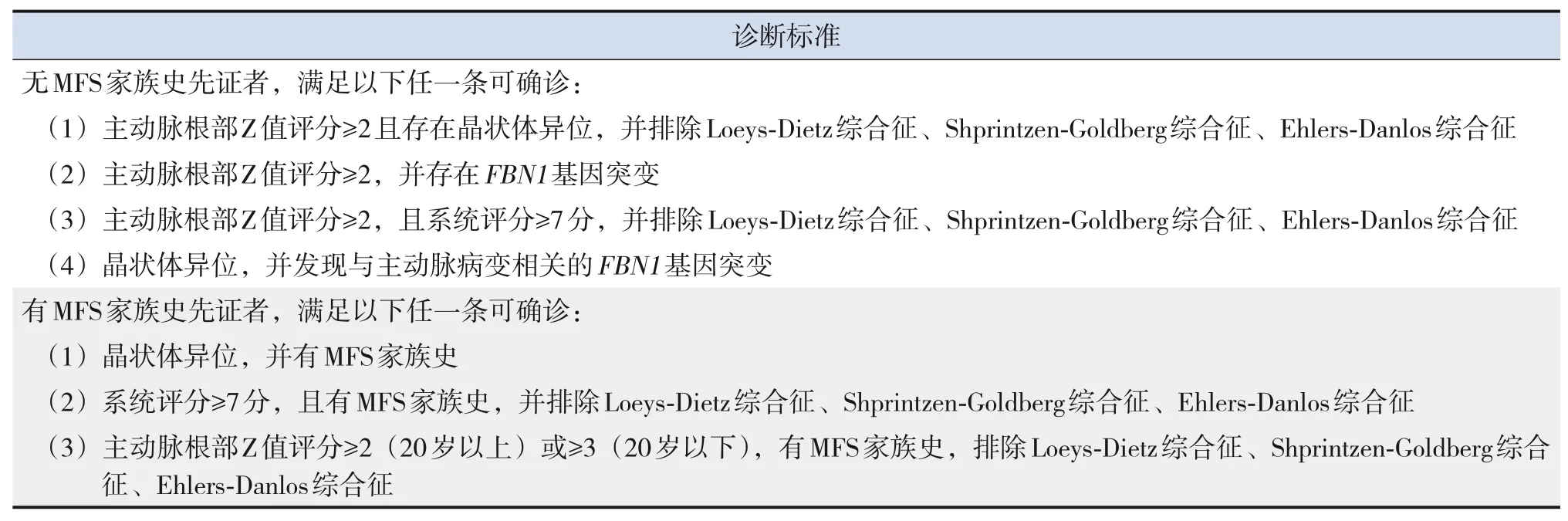
表1 修订版根特标准
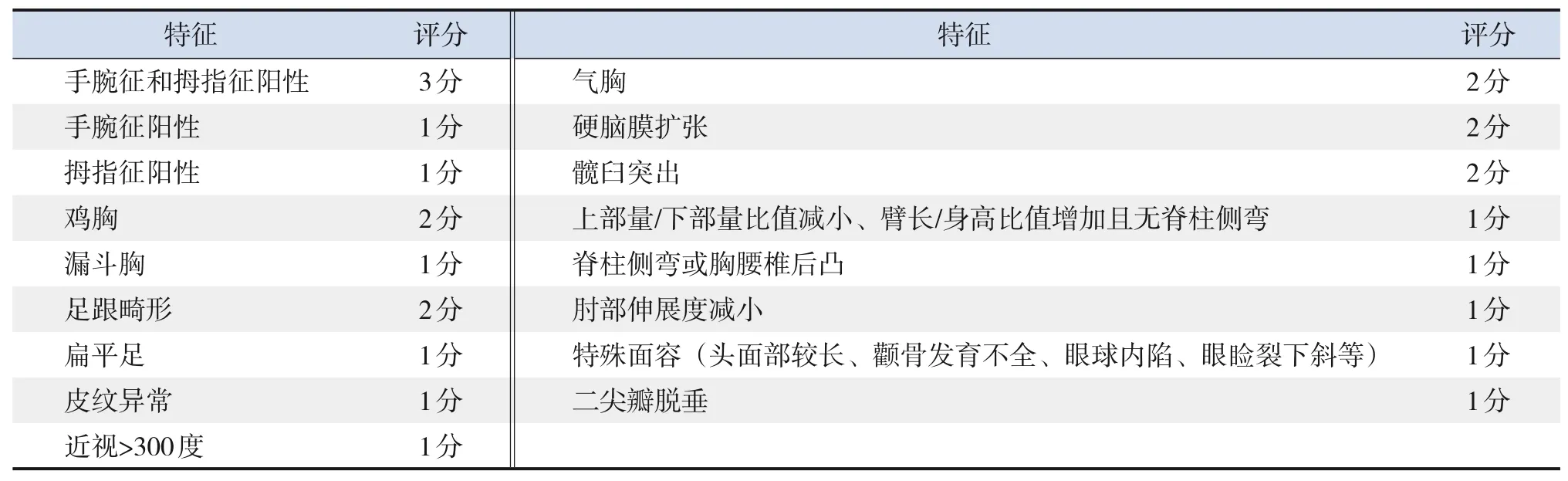
表2 MFS的系统特征评分
2 MFS的治疗
2.1 药物治疗
心血管疾病是MFS 患者的主要致死因素,如主动脉夹层或破裂、主动脉瘤、心脏瓣膜脱垂、心内膜炎、心肌病、心力衰竭等。MFS 的药物治疗旨在限制主动脉的扩张速度、延缓心血管疾病进展,无法达到治愈目的,药物治疗仍是目前MFS的研究热点。
β受体阻滞剂能降低主动脉近端血流动力学压力、延缓主动脉根部扩张,被推荐为MFS 的一线治疗药物,普萘洛尔或阿替洛尔广泛用于儿童,美托洛尔和比索洛尔常用于成人[21-22]。但2017 年一项系统评价指出尚缺乏确凿证据证明β受体阻滞剂在降低MFS 病死率及主动脉相关手术率方面的有效性,且具有一定不良反应,β受体阻滞剂作为预防MFS 患者主动脉并发症的主要疗法仍有很大挑战[23]。
与单独使用β 受体阻滞剂治疗的MFS 患者相比,加用血管紧张素Ⅱ受体阻滞剂(angiotensin Ⅱreceptor blocker,ARB)可进一步预防或减轻主动脉壁的病理进展、减缓主动脉根部扩张,并能够减少已行主动脉根部置换术患者的主动脉弓部扩张,目前报道的可用于治疗儿童MFS 的ARB 类药物有氯沙坦、厄贝沙坦,但需要大型临床试验进一步证实[24-27]。研究[28]指出,在小鼠模型中使用血管紧张素受体脑啡肽酶抑制剂比单独使用ARB更能延缓升主动脉扩张,MFS 患者中首例使用沙库巴曲缬沙坦的病例报告表明,这种治疗可改善MFS 相关的心肌病,血管紧张素受体脑啡肽酶抑制剂治疗可能是一种新的MFS治疗方法。
血管紧张素转化酶抑制剂(angiotensin converting enzyme inhibitor,ACEI)具有降低动脉压力、延缓动脉硬化的作用,研究发现FBN1基因突变能增强TGF‐β 的信号转导作用,而ACEI 类可降低TGF‐β的信号转导作用,因此被推荐用于MFS患者的治疗,钙通道拮抗剂可促进血管重塑、改善血管内皮功能,但其治疗MFS 的安全性和有效性临床证据有限,应用时需谨慎[29-31]。
当患者对β受体阻滞剂有严重不良反应时,可考虑使用ARB 或ACEI、钙通道阻滞剂等药物替代[32]。2021 年Deleeuw 等[28]指出,基质金属蛋白酶抑制剂、催产素受体拮抗剂、丝裂原活化蛋白激酶级联抑制剂、γ‐氨基丁酸等可能是未来MFS的药物治疗靶点,心血管疾病的斑马鱼模型、人类诱导多能干细胞MFS 体外模型等研究可帮助确定新型药物的疗效及安全性,MFS 未来的药物研究充满了机遇与挑战。
2.2 手术及其他治疗
2.2.1 手术治疗 “Bentall‐de Bono”主动脉根部置换术的创立使MFS 患者的中位生存年龄得到了提高,目前该手术已多次改良[33]。当患者升主动脉在主动脉窦水平的直径>4.5 cm 时,建议预防性手术治疗。主动脉直径/体表面积<2.75 cm/m2的患者被认为发生主动脉夹层的风险较低,2.75~4.25 cm/m2的患者发生主动脉夹层的风险为中度,>4.25 cm/m2则为高风险,如有主动脉夹层家族史、主动脉扩张速率>5~10 mm/年或左心室扩张导致严重的主动脉瓣关闭不全等情况,可考虑更早手术干预[34]。
在nMFS患儿中,严重瓣膜功能不全可造成充血性心力衰竭,药物治疗效果不佳时,尽早考虑手术修复或置换二尖瓣、三尖瓣,但长期预后不详,且三尖瓣置换术后血栓发生率较高,心脏移植可能是nMFS患儿的最终选择[7,14]。
主肺动脉(main pulmonary artery,MPA)扩张是较主动脉扩张更严重的血管和结缔组织受累标志,MFS患者需定期检查MPA情况。Stark等[35]回顾性研究了135 名MFS 患儿,发现MPA 扩张者其主动脉扩张、二尖瓣脱垂及晶状体异位、全身多脏器受累的发生率高于无MPA 扩张患儿。MPA 扩张较常见的合并症为肺动脉瘤,MPA 夹层者较少见。
对于新生儿期发现FBN1基因异常但尚无临床表现者,平时需避免剧烈哭吵及活动,定期行超声心动图检查,根据体表面积评估主动脉内径Z值情况,并记录主动脉扩张速率,确诊MFS 或发现主动脉开始扩张后,可开始口服β受体阻滞剂,监测药物不良反应,常规半年至1年进行1次影像学检查,若主动脉根部Z值≥3或主动脉扩张速率>5~10 mm/年或出现心血管系统临床症状时,应加强随访频率,并可预防性手术治疗,这部分患儿若突变位点位于24~33 号外显子内,预后可能较差,若位于其他区域,其预后目前尚不完全明确[7,36]。
2.2.2 其他治疗 晶状体异位是MFS 患者眼部受累的常见表现,早期手术干预可避免青光眼等远期致盲并发症的发生,Sahay等[37]报道了一种显微镜引导下的晶状体抽吸技术,用于治疗儿童晶状体前脱位。
约60%的MFS 患者存在脊柱侧弯,严重时可致明显的骨骼畸形、疼痛、限制性通气功能障碍。其严重程度通过测量Cobb 角评估,当Cobb 角<25°,建议姿势矫正训练并定期随访;当25°≤Cobb角≤40°且无明显症状时,可予支具治疗;若Cobb角>40°或引起症状时,建议手术治疗[38-39]。
3 MFS产前诊断及干预
对MFS 高风险妊娠家庭需进行产前诊断或胚胎植入前基因检测。nMFS 的产前诊断多基于心血管系统的异常,Wang 等[40]报道了1 例25 周的胎儿,无MFS 家族史,胎儿心脏超声示多瓣膜发育不良、心脏扩大,股骨超声发现其长度大于同胎龄的+2 SD,引产后脐血基因检测为FBN1基因新发突变,确诊nMFS,作者进一步指出与nMFS 相关最常见的心血管异常包括心脏扩大、瓣膜返流、主动脉和/或肺动脉根部扩张、瓣膜脱垂等,当心脏扩大严重,导致充血性心力衰竭、肺部体积受压,严重时造成患儿死亡。
Veiga‐Fernández等[41]也指出nMFS最常见的产前检查结果是心血管异常,若产前检查发现心脏问题,应进一步检查其他部位有无异常。nMFS 患儿生后15个月内的病死率约73.68%,早发性MFS的产前诊断具有很大的挑战性,需多学科参与。
Wang 等[42]运用胚胎植入前遗传学诊断(pre‐implantation genetic diagnosis,PGD)的方法帮助一对夫妇(父亲为MFS 患者)产下一个无致病基因突变、无先天性心脏畸形的新生儿,截至目前已有多项报道[43-44]运用PGD 方法筛选胚胎中的致病突变,除MFS 外,PGD 方法也可用于其他单基因遗传病,结合三代测序技术,进一步为具有遗传病高风险的家庭获得健康的后代。
4 小结
MFS 可累及全身多个系统,临床表现不一,不典型患者诊断时需与其他结缔组织疾病相鉴别,确诊后建议尽早口服药物干预,密切随访心脏、大血管情况,必要时手术干预。nMFS 患儿生后即发病,进展快,病情重,早诊断早治疗有助于改善患儿预后。评估主动脉受累程度、明确基因型-表型关系将对创建更准确的风险分层系统、建立有效的遗传咨询及个性化诊疗方案具有重要意义。产前遗传诊断对于MFS 高遗传风险的家庭意义重大,需加强这部分家庭的产前咨询意识。关于MFS 的治疗,建议由包括临床遗传学家、心脏病学家、眼科医生、骨科医生和心胸外科医生等在内的多学科团队进行综合管理,为延长MFS 患者的生存年限、提高其生活质量而努力。
