5-氯代噁唑并[4,5-b]吡啶-2-胺的简便合成
2022-07-22徐小娜张婧王晓霞仝红娟朱周静
徐小娜,张婧,王晓霞,仝红娟,朱周静
(1.咸阳职业技术学院医药化工学院,陕西 咸阳 712000;2.咸阳市分子影像与药物合成重点实验室/陕西国际商贸学院医药学院,陕西 咸阳 712046)
杂环结构普遍存在于自然界以及人工合成分子中,具有广泛的生物活性[1-3]。目前,已经上市的药物及农药分子结构中,很多都含有杂环结构[4-5]。其中,噁唑类及吡啶类杂环化合物是两大类具有代表性的杂环化合物,相关的合成及活性研究已有大量的文献报道[6-8]。该类化合物具有广泛的生物活性,如RSK2抑制活性[9],腺苷A3受体激动活性[10],抗锥虫病活性[11],极光激酶抑制活性等[12]。
噁唑并[4,5-b]吡啶是一种特殊的杂环化合物母核,分子中同时含有噁唑环和吡啶环结构,其合成及活性研究已成为当前药物化学研究的热点课题之一。随着药物及合成化学的快速发展,噁唑并[4,5-b]吡啶化合物在药物化学中的作用将会日益重要[13-15]。目前,关于噁唑并[4,5-b]吡啶类化合物的合成,主要方法有2种:(1)2-氨基吡啶-3-醇衍生物与二硫化碳在乙醇中回流反应,得到的产物再与二氯亚砜反应制备得到2-氯噁唑并[4,5-b]吡啶,该方法需要2步反应,用到的二硫化碳和二氯亚砜对环境污染较大[10];(2)2-氨基吡啶-3-醇衍生物与苯甲酸衍生物反应制备得到2-苯基噁唑并[4,5-b]吡啶,该方法缺点是以毒性较大的二甲苯为溶剂或者强酸PPA(多聚磷酸)等作为催化剂,对环境造成较大污染[11,16]。作为噁唑并[4,5-b]吡啶类化合物中的一种,5-氯代噁唑并[4,5-b]吡啶-2-胺母核的合成研究目前尚未见文献报道。因此,探索5-氯代噁唑并[4,5-b]吡啶-2-胺母核的合成方法,然后以此母核为基础,开发结构新颖的噁唑并[4,5-b]吡啶类化合物用于活性筛选,在药物化学领域中具有一定的研究意义。近年来,绿色合成的理念得到了广泛的重视,以水为反应溶剂是绿色合成的重点研究方向之一[17]。
为了能开发一种合成路线简单、低成本、条件温和的合成5-氯代噁唑并[4,5-b]吡啶-2-胺(1a)的方法,进而为研究噁唑并[4,5-b]吡啶类抗肿瘤活性药物分子中的开发提供母核结构。参考文献[10-11]以2-氨基吡啶-3-醇为原料合成噁唑并[4,5-b]吡啶母核的策略,本研究以2-氨基-6-氯吡啶-3-醇(2a)为原料,水为反应溶剂,与BrCN一步反应合成得到5-氯代噁唑并[4,5-b]吡啶-2-胺(1a)(合成路线见图1),并考察了影响1a收率的主要因素,确定适宜的反应条件。该工艺条件同样适用于5-溴代噁唑并[4,5-b]吡啶-2-胺(1b)的合成。应用研究发现,化合物1a或1b与苯硼酸频哪醇酯发生Suzuki偶联反应,可以用于制备5-苯基噁唑并[4,5-b]吡啶-2-胺(3)。该合成方法以水为反应溶剂,成本低、反应条件温和,具有一定的实际应用价值。

图1 5-氯/溴代噁唑并[4,5-b]吡啶-2-胺(1a,1b)的合成路线Figure 1 The synthetic route for 5-chloro/bromine oxazolo[4,5-b]pyridin-2-amine(1a,1b)
1 实验部分
1.1 仪器与试剂
APEX II CCD型X线单晶衍射仪(德国Bruker公司);AV400型核磁共振仪(DMSO-d6为溶剂,TMS为内标,德国Bruker公司);Ultima Global Spectrometer型质谱仪(ESI源,美国Waters公司);XT4型显微熔点测定仪(温度计未校正,北京科仪电光仪器厂);RE-52AA旋转蒸发仪(上海亚荣生化仪器厂);SHB-Ⅲ型循环水式多用真空泵(郑州长城工贸有限公司)。
2-氨基-6-氯吡啶-3-醇、2-氨基-6-溴吡啶-3-醇、BrCN、苯硼酸频哪醇酯(阿达玛斯试剂有限公司);柱层析硅胶(300~400目,青岛海洋化工厂);其他试剂均为市售分析纯。
1.2 5-卤代噁唑并[4,5-b]吡啶-2-胺(1a,1b)的合成
在100 mL的三口瓶中,依次加入2-氨基-6-氯/溴吡啶-3-醇(2a,26.2 mmol)、4.16 g(39.3 mmol)BrCN和50 mL水,在70℃下搅拌3 h,TLC监测反应结束后,体系降至室温,加入饱和NaHCO3水溶液调节至碱性(pH=10),用乙酸乙酯萃取(3×40 mL),分离有机相,经无水Na2SO4干燥、减压浓缩,所得粗品经硅胶柱层析分离纯化(二氯甲烷-甲醇,体积比15∶1],得到目标化合物(1a、1b)。
5-氯代噁唑并[4,5-b]吡啶-2-胺(1a):淡黄色固体,收率86.5%。1H NMR(400 MHz,DMSO-d6)δ:8.21(brs,2H),7.64(d,J=8.0 Hz,1H),7.14(d,J=8.0 Hz,1H);13C NMR(100 MHz,DMSO-d6)δ:166.1,159.4,140.5,134.3,118.5,118.0。ESIMS,m/z:170.01[M+H]+。
5-溴代噁唑并[4,5-b]吡啶-2-胺(1b):黄色固体,收率 81.9%。1H NMR(400 MHz,DMSO-d6)δ:8.23(brs,2H),7.65(d,J=8.0 Hz,1H),7.15(d,J=8.0 Hz,1H);13C NMR(100 MHz,DMSO-d6)δ:166.0,159.8,141.1,134.8,119.1,118.8。ESIMS,m/z:214.05[M+H]+。
1.3 5-苯基噁唑并[4,5-b]吡啶-2-胺(3)的合成
无氧条件下,将(2.3 mmol)化合物1a或1b溶于DMF(5 mL)中,再加入 636 mg(4.6 mmol)K2CO3、H2O(0.5 mL)、714 mg(3.5 mmol)苯硼酸频哪醇酯,搅拌均匀后再加入146 mg(0.2 mmol)Pd(dppf)Cl2,在100℃下反应3 h,TLC监测反应结束后,加入饱和食盐水(20 mL)稀释,乙酸乙酯萃取(3×20 mL),有机相经无水Na2SO4干燥后减压浓缩,粗品通过硅胶柱层析分离纯化(二氯甲烷-甲醇,体积比20∶1),得到黄色固体。当以1a为反应原料发生Suzuki偶联反应,产物3的收率为62.2%;当1b为反应原料发生Suzuki偶联反应,产物3的收率为78.6%。1H NMR(400 MHz,DMSO-d6)δ:8.05-7.96(m,4H),7.73(d,J=8.2 Hz,1H),7.53(d,J=8.2 Hz,1H),7.46(t,J=7.5 Hz,2H),7.38(t,J=7.3 Hz,1H);13C NMR(100 MHz,DMSO-d6)δ:165.6,159.0,151.4,140.3,139.7,129.0,128.6,126.8,115.7,112.7。LC-MS[M+H]+m/z:211.11。
2 结果与讨论
2.1 化合物1a的合成
以2-氨基-6-氯吡啶-3-醇(2a)与BrCN反应合成5-氯代噁唑并[4,5-b]吡啶-2-胺(1a)的反应为模型,研究反应条件对产物(1a)收率的影响,筛选最佳反应工艺条件。
2.1.1 反应溶剂对化合物1a收率的影响 按“1.2”项下反应条件,考察反应溶剂(甲醇、乙醇、水、N,N-二甲基甲酰胺)对产物1a收率的影响,结果发现:反应在4种溶剂中均可进行,其中,在甲醇和乙醇中,产物1a收率较低,分别为23.7%和32.0%,而在水中和N,N-二甲基甲酰胺2种反应溶剂中收率较高,分别为86.5%和55.2%,以水作为反应溶剂时的产物收率最高。因此,确定反应溶剂为水。
2.1.2 物料比对产物1a收率的影响 按“1.2”项下反应条件,考察反应中物料比即n(BrCN)∶n(2a)对产物1a收率的影响,考虑原料成本因素,选择BrCN用量过量。研究发现:当物料摩尔比n(BrCN)∶n(2a)=1.1∶1时,收率仅为33.9%;提高BrCN用量,产物1a收率提高,当BrCN用量增加至摩尔比n(BrCN)∶n(2a)=1.5∶1时,收率最高达到86.5%,而继续增加BrCN用量分别为1.6∶1和1.7∶1时,收率基本保持不变。因此,确定适宜的物料摩尔比为n(BrCN)∶n(2a)=1.5∶1。
2.1.3 反应温度和时间对产物1a收率的影响 在确定了反应溶剂及适宜物料比后,按“1.2”项下反应条件,考察反应温度(20、50、70、90、100℃)对产物1a收率的影响。结果发现:在20℃条件下,产物1a收率为36.5%;继续升高反应温度,产物1a收率逐步升高,当反应温度升高到70℃,产物1a收率最高达到86.5%;继续升高反应温度至100℃,产物收率开始降低。因此,确定适宜反应温度为70℃。
按“1.2”项下反应条件,考察反应时间(1、2、3、4 h)对产物1a收率的影响。结果发现:当反应1 h时,产物1a收率较低,仅为43.3%;反应2 h时,产物1a收率增加至77.6%;当反应3 h时,产物1a收率最高,达到86.5%;继续延长反应时间,收率没有明显变化。因此,确定适宜的反应时间为3 h。
2.2 化合物1a的晶体结构
2.2.1 化合物1a晶体结构测定 将化合物1a溶解于乙醇溶剂中,室温自然挥发,大约3 d后得到淡黄色晶体,选取尺寸为0.12 mm×0.08 mm×0.05 mm的单晶,在APEX II CCD型X线单晶衍射仪于173 K条件下,收集数据采用石墨单色化的MoKα射线(λ=0.071 073 nm),用ω-2θ扫描方式,衍射指标范围为-23≤h≤23,-24≤k≤18,-19≤l≤19,在θ角的范围为2.658°<θ<26.384°,收集到衍射点14 031个,独立衍射点3 080个 [R(int)=0.057 5],数据的处理采用SHELXL-97程序直接解出晶体结构,最终得到R1=0.041 4,ωR2=0.093 9,残余电子密度峰最大值435 e/nm3,最小值-357 e/nm3。化合物1a的晶体及精修参数如表1所示。
2.2.2 化合物1a晶体结构分析 化合物1a晶体(CCDC号:2006787)结构属于四方晶系,I41/a空间群,晶胞参数a=1.966 41(8)nm,b=1.966 41(8)nm,c=1.561 44(5)nm,α=β=γ=90.00°,V=6.037 7(5)nm,Z=16,Dc=1.572 g/cm3,μ=0.455 mm-1,F(000)=2912.0,见表1。通过X-ray单晶衍射可以看到化合物1a的立体结构,图2为分子结构图。表2和表3分别为化合物1a的键长和键角数据。

表1 化合物1a的晶体及精修参数Table 1 Crystal structure and refinement parameters for compound 1a

表2 化合物1a的键长Table 2 Bond lengths for compound 1a
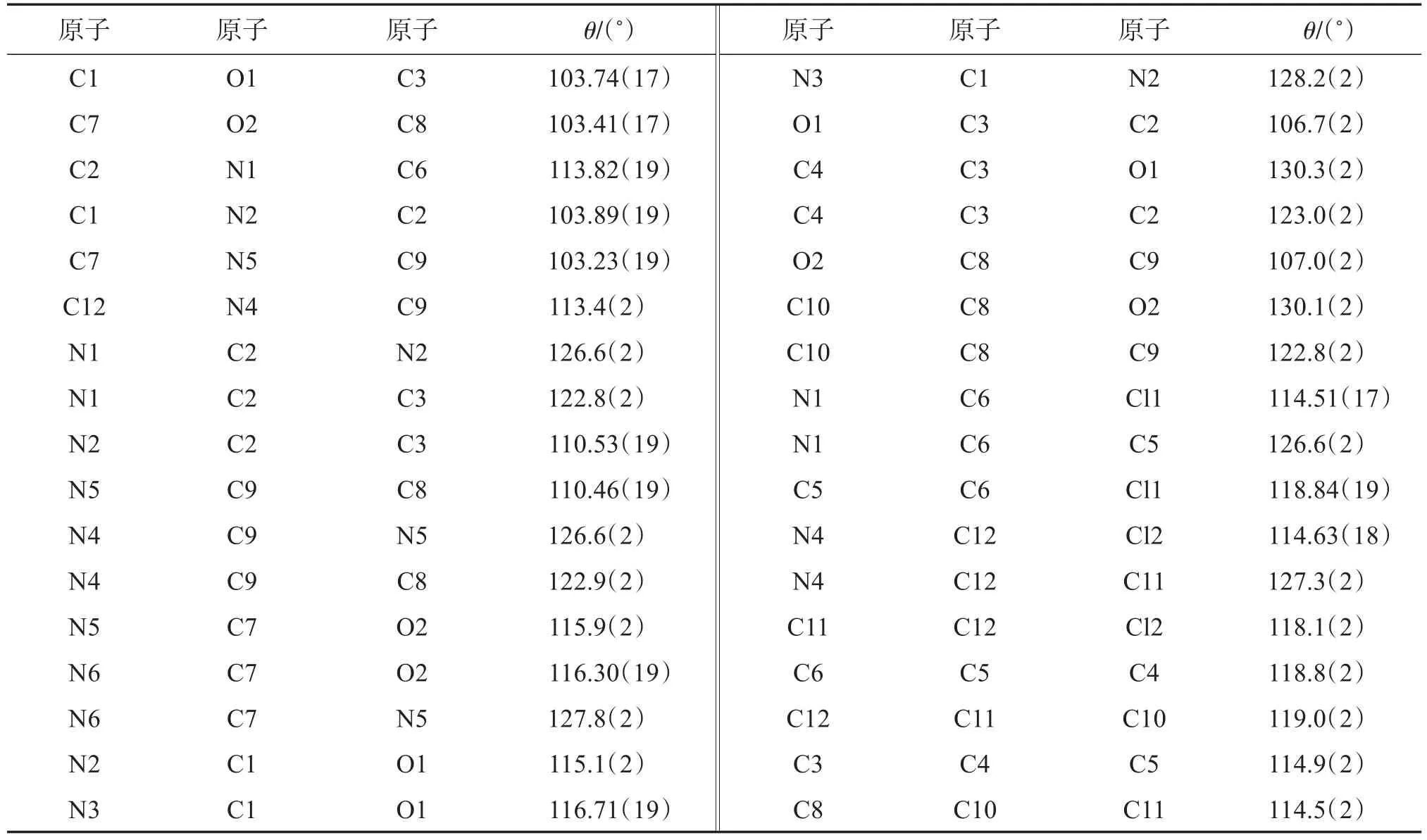
表3 化合物1a的键角Table 3 Bond angles for compound 1a
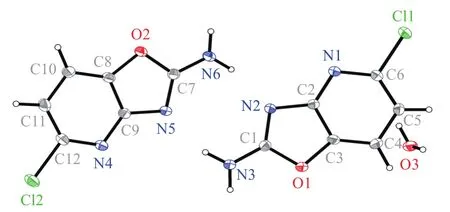
图2 5-氯代噁唑并[4,5-b]吡啶-2-胺(1a)的晶胞结构图Figure 2 Crystal cell structure of 5-chlorooxazolo[4,5-b]pyridin-2-amine(1a)
2.3 5-苯基噁唑并[4,5-b]吡啶-2-胺(3)合成
为了进一步研究5-氯/溴代噁唑并[4,5-b]吡啶-2-胺(1a、1b)在有机合成方面的应用,1a或1b与苯硼酸频哪醇酯发生Suzuki偶联反应,可以制备5-苯基噁唑并[4,5-b]吡啶-2-胺(3),收率分别为 62.2%和78.6%。化合物3结构经1HNMR、13C NMR和ESI-MS表征,合成路线见图3。
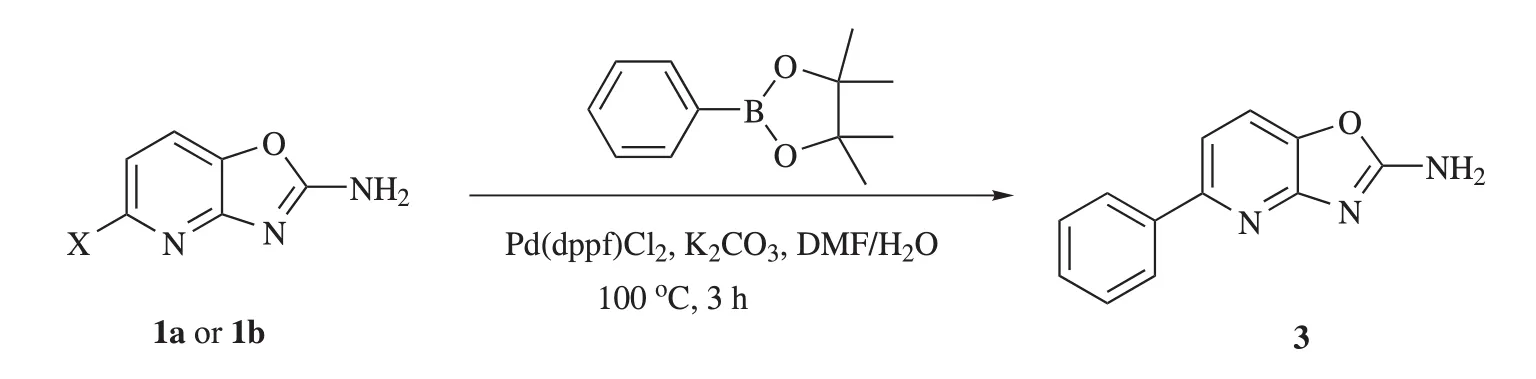
图3 5-苯基噁唑并[4,5-b]吡啶-2-胺(3)的合成路线Figure 3 The synthetic route of 5-phenyloxazolo[4,5-b]pyridin-2-amine(3)
3 结论
本文首次报道5-氯代噁唑并[4,5-b]吡啶-2-胺(1a)的合成方法,该方法以2-氨基-6-氯吡啶-3-醇(2a)和BrCN为原料,在水溶剂中,一步反应得到目标产物。该合成方法以水为溶剂,不仅反应成本低、绿色环保,而且产物收率较高,具有一定的应用价值。本研究采用氨基吡啶来构建噁唑并[4,5-b]吡啶衍生物,为构建杂环化合物提供了一种新的选择。另外,产物1a、1b经过Suzuki偶联反应可以用于构建5-芳基取代的噁唑并[4,5-b]吡啶-2-胺衍生物。目前,关于该Suzuki偶联反应的条件优化、底物范围拓展及生物活性研究等工作正在进行中。
