基于DNA 甲基化鉴定口腔鳞状细胞癌预后生物标志物
2022-07-18王婷婷温凌杜王子弘李孔亮古妍琪杨宏宇
王婷婷,温凌杜,王子弘,李孔亮,古妍琪,杨宏宇
(1.北京大学深圳医院口腔医学中心,广东 深圳 518000;2.深圳市宝安妇幼保健院口腔科,广东 深圳 518000;3.南方医科大学公卫学院,广东 广州 510000)
口腔鳞状细胞癌(oral squamous cell carcinoma,OSCC)是口腔颌面部最常见的癌症之一,每年影响约超过40 万人[1]。尽管近几十年来诊断和治疗方法不断更新,但由于OSCC 具有高复发率和高颈部淋巴结转移的风险,导致OSCC 的预后并没有明显改善,5 年总生存率仍为45%~50%[2]。因此,有必要进一步阐明OSCC 的发病机制,以挖掘潜在的生物标志物以改善预后。DNA 甲基化是表观遗传修饰的重要表现形式,可影响血管生成、细胞周期的调控或DNA 损伤修复等多个方面[3],而异常的DNA 甲基化与肿瘤发病机制和癌症患者的生存率显著相关[4]。因此,本研究应用生物信息学方法分析OSCC 样本的DNA 甲基化高通量微阵列信息,并分别进行基因本体论(gene ontology,GO)和京都基因和基因组百科全书(kyotoencyclopedia of genes and genomes,KEGG)通路富集分析、蛋白质-蛋白质相互作用(protein-protein interaction,PPI) 网络和Cytoscape软件的MCODE 模块分析以及生存分析,以期鉴定出OSCC 差异甲基化区域(differentially Methylated Region,DMR)中与预后相关的关键基因,并初步探究其在OSCC 中的作用机制,现报道如下。
1 资料和方法
1.1 数据下载与整理 在GEO 数据库中以“Methylation,450k,OSCC”为检索条件,筛选并下载DNA 甲基化分析数据集GSE87053,平台信息为GPL13534 IlluminaHumanMethylation450 BeadChip (Human-Methylation450_15017482),共包含21 个样本,其中正常样本10 例,OSCC 样本11 例。
1.2 数据处理与DMRs 的筛选 应用R 软件(V 3.6.1)minfi、impute、wateRmelon、IlluminaHumanMethylation450kmanifest、IlluminaHumanMethylation450kanno.ilmn12.hg19 和cluster 包对数据行归一化处理及质控,进一步以qvalue <0.05 作为具有统计学差异的阈值筛选出差异甲基化位点(differentially methylated position,DMP),最后以“cutoff=0.2,B=100,type=Beta”为设置条件,以fwer<0.05 作为具有统计学差异的阈值,将DMPs 聚类以得出DMRs。
1.3 DMRs 的基因注释 将DMRs 数据提交至wANNOVAR,以注释出DMRs 中所包含的基因。
1.4 GO 与KEGG 通路富集分析 为明确DMRs 中所包含基因的生物学特性,通过DAVID 数据库行GO和KEGG 通路富集分析,其中GO 富集分析包括细胞组分(cellular component,CC)、分子功能(molecular function,MF) 和生物过程(biological process,BP)。结果均以Pvalue<0.05 为有统计学差异。
1.5 PPI 网络的构建与关键基因的鉴定 为在蛋白质水平上进一步探索DMRs 中所包含基因间的关联,将DMRs 中所包含的基因映射到STRING 数据库中,并同时去除无关联基因以及将interaction score≥0.4 作为阈值,来评估这些基因间的相互作用关系。进一步应用Cytoscape 软件(V 3.6.1)中的MCODE(V 1.6.1)模块,以“degree cutoff=2,max.Depth=100,k-core=2,node score cutoff=0.2”为设置条件,对PPI 网络进行聚类以发现密集的连接区域,并鉴定出PPI 网络中的关键基因。
1.6 OSCC 关键基因的预后相关性分析 通过GEPIA数据库行OSCC 关键基因的Kaplan-Meier 分析,明确关键基因表达与OSCC 患者总生存期(overall survival,OS)的相关性。以P<0.05 为差异有统计学意义。
2 结果
2.1 DMPs 与DMRs 的筛选 由图1 可见,GSE87053数据集经过R 软件minfi、impute 等包处理后Beta中位值位于同一水平,并筛选出cg05068636、cg17477990 和 cg13260377 等 93 650 个 DMPs(qvalue <0.05),见表1;聚类得出chr6(start 31696223~end 31696729)、chr2(start 54086854~end 54087343)和chr1(start 25257624~end 25258146)等168 个DMRs(fwer<0.05)见表2。
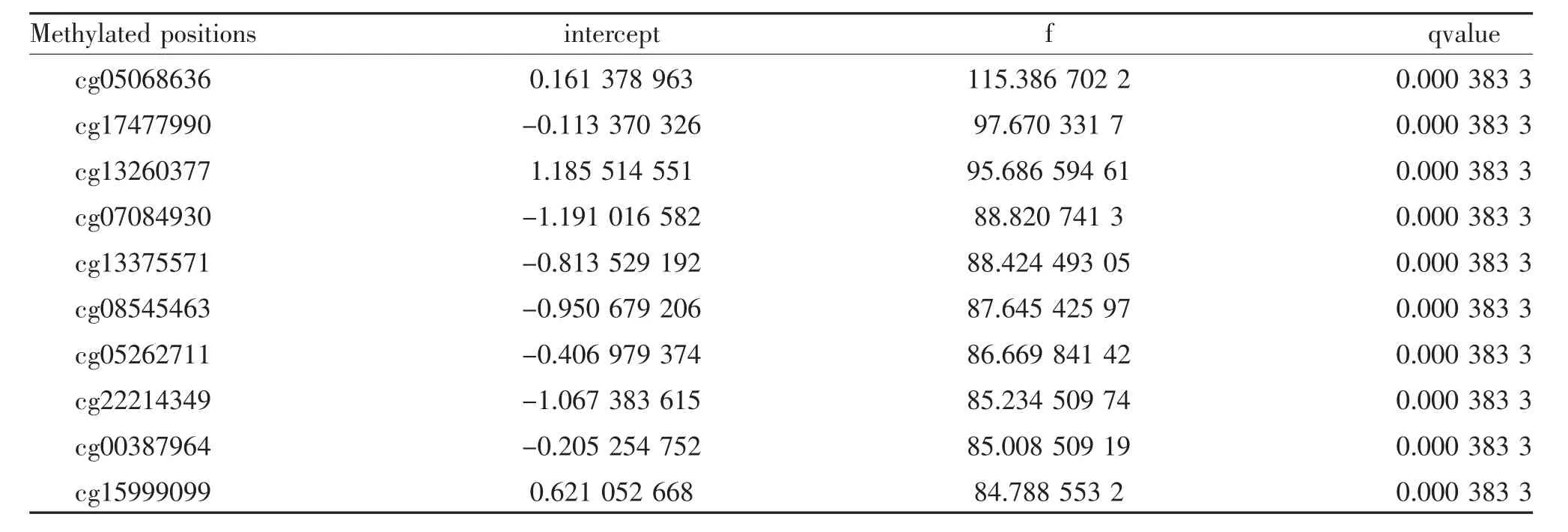
表1 差异甲基化位点前10 位
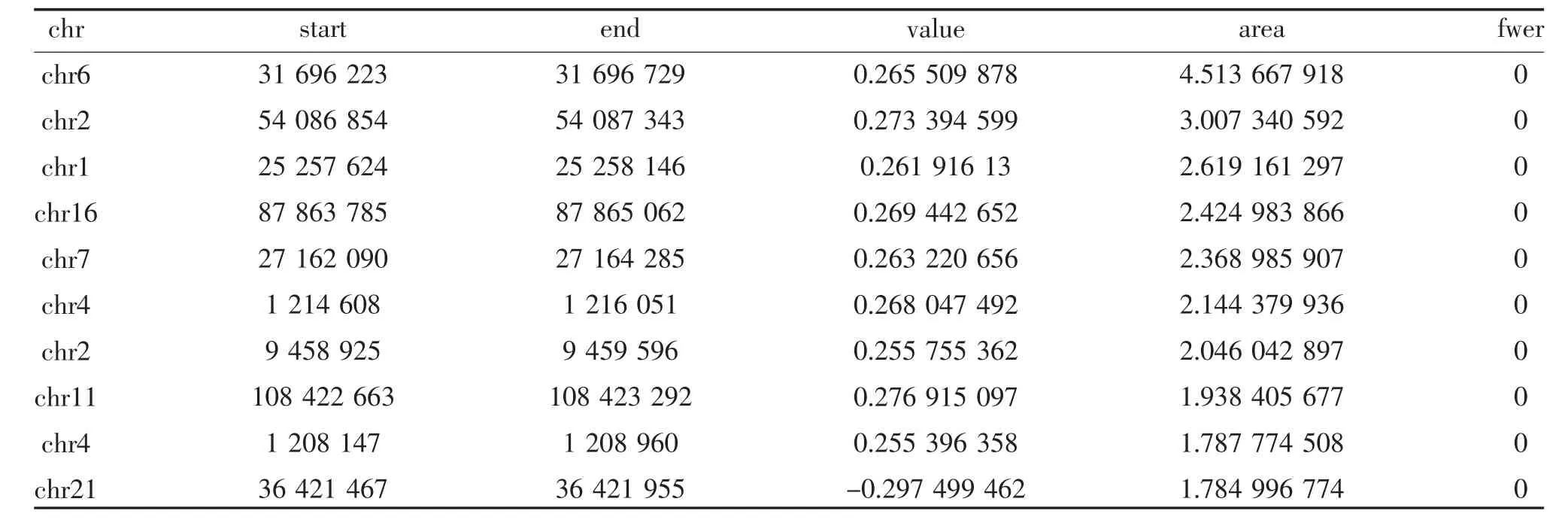
表2 差异甲基化区域前10 位
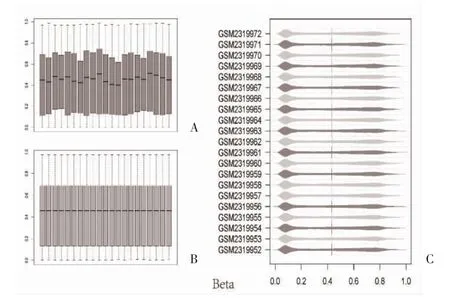
图1 数据归一化处理及质控
2.2 DMRs 的基因注释 将168 个DMRs 数据提交至wANNOVAR 工具中,共注释得到194 个基因信息,见表3。
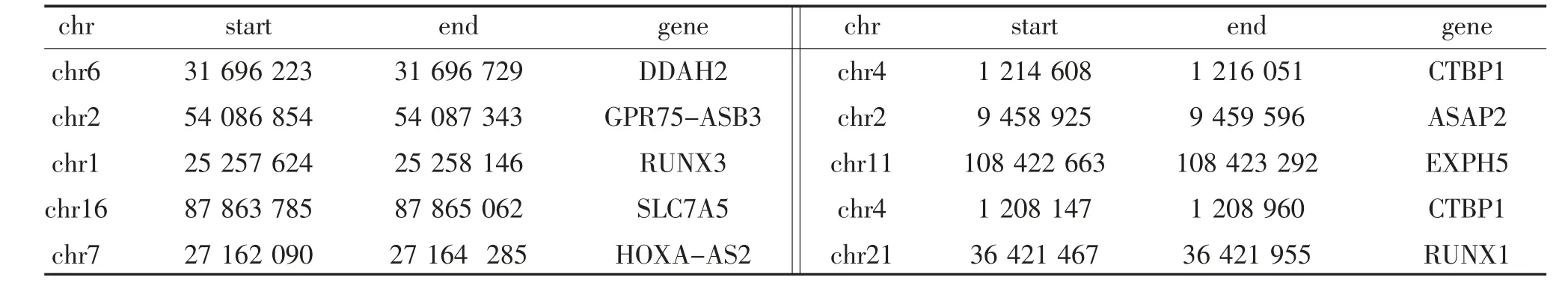
表3 其中10 位差异甲基化区域的基因注释
2.3 GO 与KEGG 通路富集分析 为阐明DMRs 中包含的194 个基因的生物学特性,通过DAVID 数据库行GO 与KEGG 通路富集分析。GO 分析结果表明(P<0.05),BP 的变化在转录、DNA 模板化和RNA 聚合酶Ⅱ启动子转录的正负调控等条目显着富集;MF 的变化主要集中在转录因子活性、序列特异性DNA 结合和氨基酸跨膜转运蛋白活性等条目;CC 的变化主要集中在转录抑制复合物和树突。KEGG 通路富集分析结果表明(P<0.05),主要富集于癌症通路、慢性粒细胞白血病、胰岛素信号通路和Notch 信号通路,见图2。

图2 差异甲基化区域基因的GO 和KEGG 通路富集分析
2.4 PPI 网络分析与关键基因的鉴定 DMRs 中包含的194 个基因的PPI 网络在去除无关联基因后,共由56 个节点和53 条边缘组成(interaction score≥0.4),见图3A。通过Cytoscape 软件的MCODE 模块聚类出具有高度连通性的连接区域(degree cutoff=2,max.Depth=100,k-core=2,node score cutoff=0.2),见图3B,可见CTBP1、RUNX1、NCOR2、CTBP2 和HDAC4 可作为OSCC 的关键基因,可能在OSCC 患者的DNA 甲基化中起重要作用。

图3 差异甲基化区域基因的PPI 网络
2.5 OSCC 关键基因的预后相关性 通过GEPIA 数据库行关键基因的Kaplan-Meier 分析,明确CTBP1、RUNX1、NCOR2、CTBP2 和HDAC4 的表达与OSCC 患者预后间的关系。结果显示,CTBP1 和HDAC4 与OSCC 患者的OS 相关(P<0.05),见图4。

图4 OSCC 关键基因的预后分析
3 讨论
既往研究已发现多种类型的表观遗传修饰,包括DNA 甲基化、组蛋白修饰和染色质重塑等,其中DNA 甲基化可通过调节细胞中基因的表达影响细胞增殖、细胞周期和细胞分化等多种生物学过程[5]。可见,DNA 甲基化的失调可能会导致肿瘤抑制因子的沉默或癌基因的表达,进而导致肿瘤的发生和发展[6]。近年来,学者们已开始通过DNA 甲基化来不断阐明OSCC 的发病机制,以寻找潜在的生物标志物。在OSCC 中已证明,LATS1 和LATS2 的甲基化失调可影响细胞周期的调控以及DDR 信号的传导[7],PAX1 和ZNF582 的高甲基化状态与肿瘤细胞的侵袭性进展相关[8]。但目前的研究尚缺乏使用数据分析来筛选和鉴定用于预测OSCC 预后的生物标志物。因此,本研究拟通过生物信息学方法分析GEO数据库中OSCC 患者的高通量微阵列芯片测序信息,以期鉴定出参与OSCC 表观遗传调控的潜在关键基因,并明确其在预后评估中的临床价值。
本研究首先基于GEO 数据库中的DNA 甲基化微阵列数据集GSE87053,分析了11 例OSCC 样本和10 例正常样本的DNA 甲基化信息,筛选出93 650 个DMPs 和168 个DMRs,通过基因注释发现DMRs 中 的 DDAH2、GPR75 -ASB3、RUNX3 和SLC7A5 等194 个的基因。GO 和KEGG 通路富集分析结果发现,这些基因可影响转录因子及其活性并参与癌症和Notch 信号通路的调节,并且已有研究表明Notch 信号可通过诱导上皮间质转化(epithelial-mesenchymal transition,EMT)从而促进OSCC 的转移[9]。可见,这些基因的生物学特性与OSCC 的发病机制密切相关。进一步PPI 网络分析显示了基因之间的功能连通性,其中CTBP1、RUNX1、NCOR2、CTBP2 和HDAC4 可作为基于DMRs 分析的OSCC关键基因,但在最后的预后分析中只有CTBP1 和HDAC4 显示出与患者OS 的相关性。
羧基末端结合蛋白1(C-terminal binding protein1,CTBP1)是转录共阻遏蛋白CTBP 的家族成员之一,可在烟酰胺腺嘌呤二核苷酸的存在下形成同源二聚体或异源二聚体,通过将HDAC、LSD1 和G9a 等表观遗传修饰酶募集到靶基因特定的启动子或增强子区域,以在致癌机制中发挥调控细胞迁移、细胞侵袭和EMT 等不同功能[10]。在胃癌中,CTBP1作为miR-539-3P 的直接靶标,其高表达可通过促进EMT 和调控肿瘤细胞的增殖和侵袭以加速疾病进展[11]。在乳腺癌中,高表达的CTBP1 还被证明可通过激活RAD51 的转录从而增加肿瘤细胞对顺铂的抗性[12]。但值得注意的是,在肺腺癌中过表达的CTBP1 与较差的总体存活率密切相关[13]。这与本研究基于GEPIA 数据库研究发现OSCC 中高表达的CTBP1 与较长的OS 相关相悖,考虑到Takayama K等[14]的研究发现在雄激素受体阳性前列腺癌细胞中CTBP1 可表现出肿瘤抑制作用,因此,CTBP1 在OSCC 中所扮演的角色仍需进一步研究阐明。组蛋白去乙酰化酶4(histone deacetylase 4,HDAC4)为IIa 类组蛋白去乙酰化酶之一,存在于转录辅抑制因子复合物中,可与组蛋白乙酰转移酶(histone acetyltransferases,HAT)控制组蛋白的乙酰化状态,在染色质的维持和功能中发挥重要作用[15]。在癌症中HAT 和HDAC4 间的平衡被改变,从而导致对细胞侵袭、细胞迁移和细胞增殖等方面的异常调节[16]。如HDAC4 mRNA 和蛋白质在食管鳞状细胞癌组织和细胞系中呈高表达,通过抑制细胞周期蛋白依赖性激酶抑制剂p21 和p27 以及上调CDK2/4 和CDK依赖性Rb 磷酸化来促进肿瘤细胞的增殖和G1/S细胞周期进程,并与较高的肿瘤分级、晚期临床分期和较差的生存率相关[17]。另外在Wang Z 等[18]研究中也发现,HDAC4 蛋白在肝细胞中过度表达且与患者的不良生存率有关。但本研究发现HDAC4 在5 年生存率上其高表达组却表现出更长的OS,与在其他类型肿瘤中的研究结果相悖,可见其在OSCC 中的调控机制仍需进研究深入探讨。
综上所述,本研究基于公开的DNA 甲基化数据行生物信息学分析发现,CTBP1 和HDAC4 或可作为OSCC 中潜在临床靶标和预后生物标志物。但尚存在一定的局限性,研究仅基于对全球公开数据库的部分数据进行分析,对于CTBP1 和HDAC4 在结合国人OSCC 患者数据中的表达以及预后情况的相关研究仍为空白区域,因此有必要进一步进行体外和体内基础实验,以更好地了解在我国OSCC患者中CTBP1 和HDAC4 的DNA 甲基化改变的影响和潜在机制,以及通过病例对照研究和前瞻性试验评估验证候选的生物标志物,但本研究中的数据或可为OSCC 潜在分子标志物和靶点的研究提供新的思路。
