预反应态模型浅析:催化活性和近过渡态分子模拟
2022-07-15SIMByuri赵一雷
SIM Byuri,赵一雷
(上海交通大学生命科学技术学院,微生物代谢国家重点实验室,上海 200240)
在合成生物学三大基本要素中,生物元件模块化和标准化涉及催化元件改造与设计[1]。例如,在重构特定代谢途径时经常调用或兼并多个跨物种天然催化元件基因;尽管长期自然进化使得天然酶在原生环境下具有高效和稳定等特点,却不易满足工程化生产的特殊需要[2]。为了解决该问题,弗朗西斯·阿诺德教授发明了定向进化策略,在比较短的时间内通过多轮优选,在天然酶的基础上构建出大量高活性突变体的文库,再从其中筛选出优势突变[3]。然而,实际应用中序列空间全突变搜索还是受到突变实验技术中密码子简并性和碱基突变偏好性等诸多问题的限制[4]。
为了提高蛋白质工程改造效率,需要结合蛋白质理性设计方法缩小有效突变实验测试范围[5]。目前蛋白质折叠原理在物理和数学上已经全部被阐明,超级计算机可以对生物大分子的微观动态行为开展高精度分子模拟,不但可以利用经典牛顿力学对中小规模的蛋白质和核酸分子力学展开毫秒甚至秒级的全原子长轨迹模拟,也可以利用量子力学对催化元件反应中心进行飞秒至皮秒级基于电子波函数的理论计算[6-7]。量子力学(QM)和分子力场(MM)模拟方法学的普及,及GPU计算机技术的发展[8],计算体系尺度逐步扩大,从传统的有机小分子到复杂金属络合物,一直到数千到数万原子级别的生物大分子体系,让我们可以对复杂生物体系的催化分子机理进行全面深入的计算模拟和系统分析,给出与实验精度媲美甚至实验上难以观察到的微观机制和反应势能面[9]。
当今超进化理性设计的瓶颈在于有限的计算资源、有限的研究时间与酶催化反应复杂势能面全原子水平飞秒精度上的接近无穷无尽计算需求之间的矛盾[10-11]。如何利用定向进化的高效突变体文库和高精度催化分子机制来推进蛋白质工程效率,成为该领域亟待解决的科学问题[12]。“预反应态”模型是基于对酶分子催化机制的深度挖掘,利用简捷的分子动力学模拟计算来阐明催化反应中心与远端氨基酸残基的相互作用[13],快速确定突变效应和底物适配性,从而进行相应的催化元件定向设计、改造和优化,体现了合成生物学催化元件的快速优化改进的工程化理念[2]。
1 预反应态模型的理论基础
随着愈来愈多的酶分子催化机制从仅仅包括活性中心的小模型,乃至在生物分子全原子、电子水平上得以阐明,酶学家们对“酶”生物催化元件为什么能够加速生化反应的理解也越来越深入[14]。20 世纪著名的理论化学家莱纳斯·卡尔·鲍林教授降低表观活化能的初步概念[15]和酶学实验数据分析中常用的米氏方程不断被改进[16-17]。现在人们已经能够在微观水平上基本理解了酶分子与底物和辅基等小分子形成米氏复合物[18],该复合物经过一系列构象转化降低了化学键断裂和形成所需的反应活化能[19]。如同有机化学合成,酶-底物复合物在催化反应历程中有众多的中间体和过渡态,酶的专一性和特异性来自这些不同能量状态复合物的统计热力学和动力学平衡[20-21]。因此突变体、底物多样性、底物浓度变化、环境温度、盐度、离子强度、酸碱度都会改变反应历程上各种中间体复合物的相对能量和布居数[22-23]。从而,即使一个在原生细胞内已经进化非常好的天然酶在实际生物合成中可能会由于底物、产物、抑制剂、激活剂浓度与原先进化环境不同而改变了催化反应势能面,从一个低活化能反应势能面转变为高活化能的势能面[24]。
如何通过氨基酸残基突变优化在新底物和新环境下高活化能反应势能面的生物合成酶?实验科学家通过多轮定向进化对氨基酸序列进行随机性的优化和改进,而理论科学家则提出了诸多酶催化模型用于生物合成酶和工业酶的理性设计和优化[25-26]。通过对反应势能面上反应物、产物、过渡态、中间体等驻点(能量极值点)的计算来透彻刻画酶催化反应机理,理论学家基本认同了酶具有将反应的总活化能(或表观活化能)降低到15~18 kcal/mol的能力,等价于50%概率发生反应的时间缩短到毫秒至秒的范围[27-28]。然而,理论计算学术界对蛋白质分子如何降低总反应活化能的机制理解上存在许多不同的看法,总结起来有如下几种:
(1)Houk 教授等[29-30]认为,类似有机化学催化反应,酶催化能力主要来自蛋白质的特殊氨基酸残基稳定了催化过程中的关键过渡态。具体来说,一个生化反应有非常多的可能途径,特定蛋白质分子的结构具有降低关键过渡态的作用,可以通过计算底物与周边关键氨基酸残基复合体Theozyme 模型来理解,然后基于所获得的过渡态与周边残基复合物的几何结构利用从内向外(inside-out)策略等构建蛋白质框架,来开发和优化新酶[31]。
(2)Warshel教授[32]提出在蛋白质的反应势能面与孤立体系计算在可比性方面存在难以克服的问题,大部分情况下酶催化反应并没有使化学键形成或断裂(化学步)的能量发生太大改变,而是通过与底物、中间体、过渡态、产物等系列小分子预组织成一个较稳定的复合物(非化学步)抵消了整个非酶反应历程中熵损失问题,从而降低了总活化能。
类似的催化理论还有Ground-state destabilization(基态活化)[33]和Transition state stabilization(过渡态稳定)[34]、Near attack conformation(近进攻构象)[35]、 Allosteric control (变 构 控 制)[36]、Dynamical effects (动态效应)[37]等等从不同角度对催化元件加速生化反应的理论阐述。
近年我们基于红霉素生物合成酶系中硫酯酶的底物释放竞争催化机制展开了较深入的理论研究,提出了在反应势能面上选择合适的“预反应态”模型可以有效地预测水解和环化途径选择,规避了生物大分子反应过渡态超高自由度复杂计算的瓶颈问题。如图1所示,我们可以从反应体系的时间尺度上理解预反应态选择在酶催化反应历程中的重要性:

图1 酶催化历程所涉及的运动时间尺度,包括了高活化能过程及多种微观运动Fig.1 Timescale in catalytic reactions,including the high-activation energy process and various motions
①酶通过调节结合能特异性捕捉周边环境中的底物分子,与底物分子(及辅因子)一起形成了一个反应物系综“米氏复合物”。这一过程与药物分子与靶标蛋白的结合过程类似[38-39],时间尺度在毫秒级以上,结合和解离的自由能势垒主要来自熵变,即从分子间随机相互碰撞中有效地进入到合适的反应区域。如果酶反应中心接近蛋白表面,结合势垒需要克服的能量比较低,与残基突变关系小,然而对于反应中心被包埋在蛋白内核的酶而且酶蛋白刚性较大时,门控作用较为明显,需要合适的突变来解除底物和酶的有效结合[40-41]。通常底物结合能的大小对应于底物浓度的高低,在原生细胞环境内,底物浓度相对较低,催化元件进化出高结合能状态有效地从周边环境中汲取底物分子。在酶活测定实验中米氏常数Km、解离常数Kd等接近原生环境中底物浓度[42]。底物与酶的结合并进入活性中心是酶催化反应的前提。
②该反应复合物系综在特定温度、特定酸碱环境等物理化学条件下发生热涨落运动,其中一些近过渡态的结构在某个瞬间大约数百飞秒时间内完成了目标化学键的断裂和形成(即所谓的化学步)[43-45]。在化学基元步的过程与其他化学反应遵循同样的物理规律,目标化学键的断裂和形成与分子内键振动的时间尺度相近,Eyring反应动力学速率方程的指前因子大约是50 fs,也代表了无反应势垒的反应发生最高速率。
③在关键化学步反应发生前,根据蛋白质运动时间尺度(图1)[13],反应复合物体系将处于一个稳定时间达纳秒以上的“预反应态”。因该状态尚不涉及后续的化学键断裂或形成,其稳定性可以利用经典分子动力学模拟计算来评估。根据从实验数据推算的催化元件表观活化能,可以估计出化学反应的发生概率50%的反应时间大概处于毫秒到秒的时间范围[46]。
由此可见,可以通过对离关键过渡态最近的预反应态在无约束分子动力学模拟轨迹中的稳定性来评估催化元件突变体的酶活及底物适配性。通过分析定向进化突变体预反应态的相对稳定性变化,结合生物信息学的序列和结构比对,可以从原子分子水平上阐明所观察到的突变效应。
2 近进攻构象
预反应态模型的前身为酶学研究中常用的近进攻构象分析(图2)。托马斯·布鲁斯等[47]在20世纪末观察到小分子体系中前线轨道布局和范德华相互作用对反应活性有显著的影响,提出了近进攻构 象 的 概 念(near attack conformation, NAC)——在活性构象中,将要形成新化学键两个原子之间的接触距离小于它们范德华半径之和,并且与过渡态状态有相近的键角。该方法把反应发生前、在最低能量构象kBT量级以内的所有可及构象(kB为玻尔兹曼常数)分为活性构象和非活性构象,而NAC 布居数则是活性构象在总构象空间的占比数。原子立体空间和经典作用会发生很大的变化,因此NAC 的布居数需要考虑影响复合物体系自由能的空间位阻效应、静电相互作用和零点能校正。鉴于当时计算机算力的限制,前期NAC 概念的发展过程中大多利用半经验方法随机搜索小分子体系数万个能量最小化的构象,例如在分子内酸催化酯降解体系,对构象集合中羧酸负离子与被进攻的酯羧基是否接近分子内酐形成的范德华接触距离进行几何结构分析从而确定近进攻态的概率。小分子体系的活性构象布居数对数值与反应速率对数值呈线性相关,而且这些活性构象的相对能量越低则反应活化能亦越小,但小分子体系里活性构象熵与反应活性不呈现相关性。
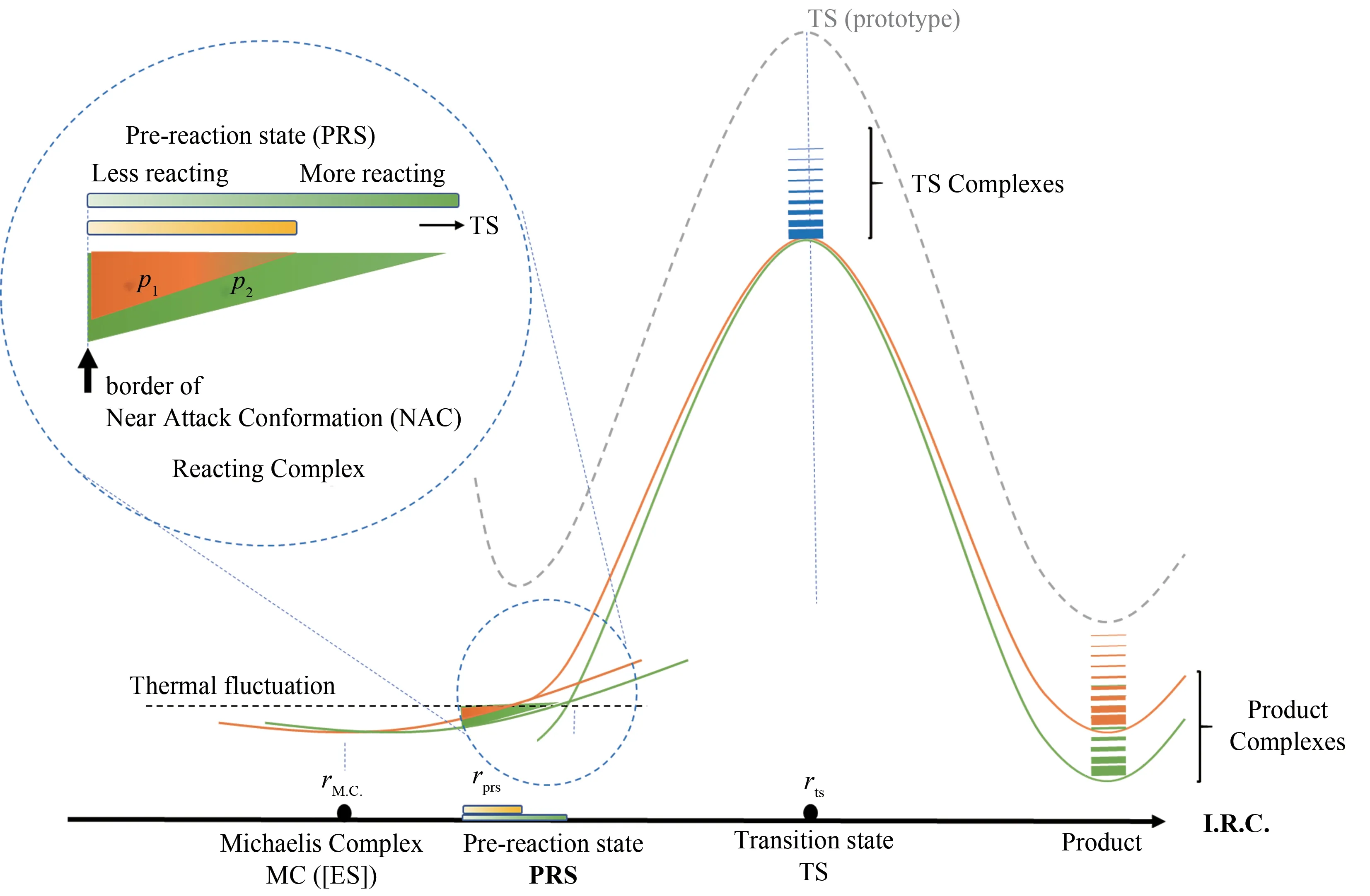
图2 催化元件突变导致反应势能面微扰的示意图[在酶催化单步反应米氏模型中,预反应态与近进攻构象定义相同,米氏复合物热运动中接近反应过渡态(近进攻构象)活性构象的布居数p与催化活性正相关]Fig.2 Diagram for the perturbation of energy profile caused by the mutation of catalytic elements(In the simplest Michaelis's model,pre-reaction state and near attack conformation are identical,using the correlation of active conformation population and enzyme proficiency.)
NAC 概念进而被推广到酶学研究中,从底物-酶的所有米氏复合物的构象中根据结构与过渡态的相似性来推断那些有利于反应发生的构象(reacting complex)。其中一个著名的NAC 例子是分支酸歧化酶的突变体活性分析[48],在水溶液中NAC 的布居数不到百万分之一,在酶和底物的米氏复合物中NAC的布居数可达30%,从而可以推算出NAC结合自由能在酶中提升了近7.8 kcal/mol,这个提升幅度接近于实验中所观察到的酶催化和非酶反应的表观活化能差(9 kcal/mol)。由于分子内张力,在分支酸异构化的计算中所选用的NAC 活性构象判定条件非常宽松,两个即将成键的碳碳原子之间距离小于0.37 nm,两个相对进攻角的正负偏差为sp2碳的法线方向20°范围。对于不同的突变体,环状过渡态的自由能势能面图计算显示高效突变体的NAC构象相对能量低,稳定性高。
布鲁斯的分支酸歧化酶计算成功验证了活性中心周边氨基酸残基与底物分子的静电和范德华相互作用可以显著提高NAC 活性构象的布居数——其原因是在催化过程中米氏复合物给反应势能面带来了结合熵陷阱,有效限制了底物的构象空间中非活性构象布居数,从而在反应势能面上转化为催化驱动力。由于酶催化反应的活化能通常低至10~15 kcal/mol,非常接近室温热运动蛋白质构象变化的能量范围,从能量的角度上过渡态与近进攻态存在某种热平衡[49]。然而,突变体自由能势能面计算需要大量的超级计算机计算资源,在蛋白质工程中对各个突变体开展自由能势能面的计算在实际应用中可望不可及,因此直接通过活性构象布居数来判断活性成为了NAC 研究的主流。在儿茶酚甲基转移酶的NAC 分析中,进攻原子碳氧之间距离定义为0.32 nm 以下,SAM 的硫碳键与碳氧夹角定义为大于165°进攻角,活性构象布居数大约为7.6%[50]。在Hhal 甲基转移酶的计算中,米氏复合物模拟轨迹中底物胞嘧啶与催化关键残基CYS18、辅酶SAM 之间的相对构象被用于判断酶活,包括了Cys17硫原子与底物胞嘧啶C6 之间的距离、SAM 甲基与底物胞嘧啶C5 之间的距离以及底物DNA 胞嘧啶碱基的N-糖苷键二面角作为3 个NAC 结构参数。由于高角度N-糖苷键二面角可以缩短进攻原子之间的距离,因此大于80°的构象被认为具有活性[51]。在甲酸脱氢酶中,NAC 定义为甲酸底物的转移氢原子与NAD 受体C4N原子之间的距离小于0.30 nm,甲酸的碳氢键与进攻原子对夹角(进攻角)在132°~180°,米氏复合物模拟中活性构象的布居数大约1.5%[52]。二氢叶酸还原酶的活性构象结构参数则定义为NADPH辅酶中被转移的负氢原子与底物DHF 的距离和夹角,在米氏复合物分子动力学模拟中两种不同手性(pro-R和pro-S)的活性构象都具有较短的距离(<0.30 nm),其中pro-R的活性构象布居数为43%,而pro-S的为7%,计算结果与实验观察到的手性选择性相吻合[53]。1,4-β-木糖酶的活性构象被用于判断广义酸碱催化二联体Glu96 和Glu177 在催化反应中的角色[54]。马肝醇脱氢酶中NAC 也被定义为氢负原子与底物的相对位置,即氢碳距离小于0.30 nm、进攻角大约132°,反应物、产物和中间体3 个体系对比中,发现pro-R质子化中间体的NAC布居数最高(约60%)[55]。在腈水解酶的近进攻构象分析中,Cys25 的硫原子与底物氰基碳原子之间的距离小于0.35 nm、进攻角设定为90°±15°,发现His159 的咪唑基对广义酸碱催化的活性构象有很大的作用[56]。在可溶性环氧水解酶中,近进攻构象定义为Asp的羧基氧原子与环氧碳原子的距离小于0.32 nm,环氧的碳氧键与进攻羧基氧原子的夹角在145°±15°范围,NAC 分析中优势活性构象与酶催化反应中环氧的两个碳原子化学选择性相吻合[57]。在嗜热吲哚3-甘油磷酯合成酶中,NAC 活性构象定义为碳碳距离0.35 nm、进攻角为120°±20°,米氏复合物在低温和高温有不同的活性构象布居数,其嗜热性通过调节活性构象的比例而实现[58]。
近进攻态概念的提出为认识酶微观催化机制提供了一个非常可行的研究工具[59-60]。Mulholland研究小组[61]从MD 模拟的16 个快照结构开始计算分支酸歧化酶催化反应路径,发现活性构象对降低活化自由能的贡献与酶对过渡态稳定化作用基本相近。而布鲁斯等[62]则把大约10%的催化作用归因于过渡状态稳定。可见,酶不但提升米氏复合物构象中的活性构象比例,而且进一步通过稳定过渡态来降低反应活化能。NAC 效应可以认为在反应开始阶段的稳定性一直保留在过渡态,例如分支酸歧化酶的静电相互作用,不但稳定了NAC 活性构象也稳定了过渡态。当然,酶催化机制还包括了动力学等其他因素[63]。
在21 世纪初,分子动力学模拟中近进攻构象和近过渡态的活性构象快照(snapshots)常作为QM/MM 过渡态计算的出发结构[64-66],随着计算机硬件的发展,更多的研究聚焦在酶催化反应势能面的构建与验证、新酶催化机制的挖掘,而对这种利用底物分子对接和半自动化分子动力学模拟可以很快地收集到的“酶活”打分函数和底物适配谱的经验性方法不甚重视[67-69]。NAC 近进攻构象的计算都是通过长时间分子动力学模拟来自底物分子直接对接的米氏复合物,从MD轨迹中统计符合玻尔兹曼分布的活性结构布居数,一旦米氏复合物中存在一个比较稳定的休止状态(resting state),或者底物-酶复合物的可及构象空间过于复杂(如生物合成中常见二元底物的催化体系),导致决速步远离米氏复合物的初始结构,从底物分子米氏复合物开始的长时间分子动力学模拟轨迹并不能高效地获得具有统计意义的活性结构布居数。
3 多步催化循环中能量模型
很久以来,计算化学家和实验者之间缺乏一个有效的话语系统,计算化学家聚焦在单步基元反应的能量势能面的变化,而生物学家则侧重表型如生物量、代谢流量、基因调控等。在蛋白质工程中则对实验数据采用了米氏方程最简单化描述方法,基于单步反应模型的米氏常数Km、最高初始反应速率vmax和催化效率kcat/Km来优化酶的催化效率。这也是导致近进攻构象分析所遇到的常见问题,即许多工业酶或生物医用酶反应并非单步基元反应,或者蛋白质工程酶分子优化设计的目标并非单底物的消耗速率而是不同产物途径的选择性问题,特别是对于多步反应过程中的中下游反应途径选择性问题,在底物和酶的米氏复合物分子模拟中并不能观察到差别。如果把活性构象分析从酶-底物的米氏复合物拓展到全反应势能面,则需要从复杂的催化循环中确定优先检测点。
近10 年来,Kozuch 从相对简单的金属有机催化循环为研究对象阐明了多基元、多底物、多进程的催化循环的能量跨度模型,以蛋白质工程量度整体催化循环效率的周转频率TOF 为目标函数[70],而非单基元反应速率[71],从催化循环的全系统考虑各个参变量变化对蛋白质催化效率的影响(图3)[72]。能量跨度模型沿用原有的表观反应活化能的概念,假设反应速率是由活性构象的浓度和Arrhenius 反应活化能决定,而活性构象的浓度或布居数是由其相对能量和稳态浓度按照玻尔兹曼分布所确定。由此可推断表观活化能的能量跨度是反应活化能和预反应态的相对能量之和,即δE为催化循环中的能量最高点和能量最低点来确定[73]。

图3 催化循环周转效率TOF与催化反应势能面的关系[在酶催化多步催化循环中,预反应态定义为关键决速步过渡态相关的活性构象的布居数p,它的变化与催化元件优化目标直接相关(如突变效应或底物适配)]Fig.3 Correlation of the turn-over frequency(TOF)and reaction potential energy surface(In multiple steps of a catalytic cycle,pre-reaction state is defined as the active conformation near rate-determining transition state,in which rate is dependent on the protein engineering target such as mutation effect and substrate diversity.)
然而,能量跨度模型在总反应能接近0的平衡中适用,如果催化循环的反应能远小于0,而一次循环的完成意味者能量的下降,新一轮的反应循环的能量参考点随即低于原有的能量零点,由此必须建立全循环周期的能量点与催化循环周转速率的关系。Kozuch 与合作者利用Eyring 方程重构了基元反应速率k空间和势能面表示方法[74],提出了整合催化反应动力学与量子力学的催化循环模型,揭示了催化循环的周转效率TOF 与反应能量校正后的能量跨度模型的关系,指出TOF 的精确表达式与催化循环势能面中过渡态和中间体的能量点贡献度,提出最高能量过渡态点和最高浓度中间体点和反应能来快速预测催化效率[75]。作者说明了催化效率TOF 的控度来自对反应速率贡献最大的状态[76],从而指示我们在量子力学计算反应势能时要重点考虑的观测点以及如何改善和调整关键基元反应步[77-78]。从优化TOF 的角度来讲,控制度接近1 的中间体和过渡态是决速中间体(TDI)和决速过渡态(TDTS)[79]。在生物体中如果一个催化元件进化得非常完善,一般而言反应物和产物之间的能量差接近于0,而且各个基元反应步的势垒接近,从能量的角度来讲达到了最优化的反应途径,导致了在该反应途径上每一个过渡态和中间体对催化转化效率都相近。反之,如果一个酶尚未进化完全,可能有若干个基元反应对催化循环的控制度超过了其他基元反应,从而使得决速中间体或过渡态的个数远远小于进化完全的催化元件(图4)。在蛋白质工程的酶进化中,更多是属于后者,当调用一个其他物种的异源基因时,大部分情况下反应途径选择在催化元件的自然进化中已经完成了,定向进化是在一个相对稳定的反应势能面内优化催化效率或对中下游的反应途径进行选择[80-82],或者是由于反应条件发生了变化,例如生物工程酶的反应物和产物的浓度远远高于自然进化催化元件时原始浓度导致反应势能面发生了相应的变化[83]。
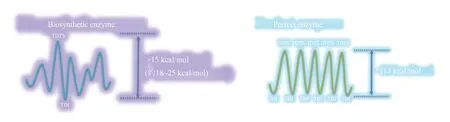
图4 催化元件分子进化前后的反应势能面[进化完成前通常存在阶段性的最大控制点(左图),而进化后反应势能面各过渡态的控制度相近(右图)]Fig.4 Imaginary reaction potential energy surface for molecular evolution(Before evolving to a"perfect"enzyme,one point on the reaction potential energy surface controls the overall rate,but many other points are equivalently important for the perfect enzyme.)
上述催化元件的势能面表述方案统一了20 世纪关于酶催化效率的基态活化、过渡态稳定、近进攻构象、动态结构、过渡态稳定等不同角度对酶催化机理的认识,把催化反应推动力和催化流与电子电路中的电势和电流概念相对应,提出了决速过渡态(TDTS)和决速中间体(TDI)协同决定催化效率,认为单个基元反应或转化状态并不具备决定催化效率的所有动力学信息。一个非常重要的推论是,TDTS 和TDI 不一定是能量最高和能量最低的状态,也不一定是某单个基元反应的相邻状态;其次,催化循环的效率并非单一决速状态,不同的目标函数对应着不同的决速参变量,在催化模型中引入了图论、相关性、反应速率控制度、鲁棒性和火山图等信息学的概念[84]。
因此,从米氏复合物的近进攻构象布居数和催化循环能量跨度模型可以延伸出可工程计算的预反应态(PRS),定义为与催化效率相关的TDI和TDTS 的预组织复合物几何结构。首先,PRS 必须是催化循环反应动力学目标函数的控制度最高的状态,它可以是米氏复合物的近进攻构象,也可能是决定多条不同产物的反应途径分岔口流量的状态[85-86]。预反应态不拘泥于米氏复合物基态、决速过渡态本身,也不拘泥于物理意义上是否必须是某种分子状态或者是某种分子状态中的“高活性”构象,或者甚至是多元底物的预组织状态,甚至是底物进入活性口袋的非化学步或是产物离开催化元件的非化学步。在分子动力学模拟中,如果该状态是与TDTS 或TDI 相邻的情况下就可以直接从活性构象的角度开始模拟测试其稳定性,如果该状态与米氏复合物相邻的情况下就使用传统底物分子对接结构开始模拟测试其可及性,如果产物和反应物的能量差接近于0,也可以适用产物分子对接结构模拟关键过渡态的预反应态,或者是反应途径的分岔口来模拟催化元件对下游不同反应途径的选择。更多的应用场景下是利用一个能量不甚准确的计算势能面,根据催化元件的进化路径分析从中选择适合计算的预反应状态,通过人工智能机器学习的方法核对该预反应态对应的控制度是否符合实验结果。
4 预反应态模型的应用
预反应态分析的基本流程为:首先,基于高阶量子力学计算提取催化中心关键过渡态的结构特征;其次,从高精度蛋白质三维结构出发,结合氨基酸质子化预测结构生物信息学工具推导出稳定存在的近进攻活性构象;最后,利用过渡态结构信息设定分子动力学模拟约束条件,通过逐步取消约束条件测试预反应态随氨基酸突变和底物变化的稳定性变化,以近进攻构象在预反应态轨迹中布居数作为“预反应态-酶活”半定量相关系数,从预反应态稳定性中挖掘酶与底物的适配图谱。
4.1 聚酮合酶系统中硫酯酶的水解与成环
聚酮合酶(PKS)是模块化多酶复合物的一个大家族[87],在药物生物合成过程中生成各种各样的环肽产物,包括大环内酯抗生素、抗真菌剂、免疫抑制剂、抗癌药物等[88]。其中红霉素DEBS合酶是Ⅰ型PKS 模块化合成的一个代表系统,由启动模块、多个延伸模块和硫酯酶组成[89-92]。硫酯酶模块化生物合成系统里负责终产物大环内酯的关环和释放[86]。实验发现底物中远离酯键的C7 位酮基改为羟基时,硫酯酶催化反应从大环内酯化反应转变为水解反应,由于关环和水解两条反应途径共享同一底物上载后的共价中间体,因此该中间体的预反应态倾向是决定水解和环化两条反应选择性的决速中间体TDI[93-94]。分子动力学模拟表明酮基和羟基两种底物与酶的共价中间体在经过10 ns模拟后都达到了平衡,在20 ns后两个体系的分子动力学模拟显示了硫酯酶lid 区呈现开合交替构象,在100 ns轨迹中则观察到了完全不同的构象演化,选择环化反应途径的底物酶共价中间体结构倾向“开”的状态,活性口袋内部体积变大,而选择水解反应途径的底物酶共价中间体慢慢倾向“闭合”构象。更为有意思的是,该硫酯酶天然底物形成的共价中间体则强烈地趋向环化反应活性结构。表明硫酯酶lid 区域通过诱导拟合互作来识别不同的底物,通过不断地“开”/“闭”构象循环调整水解/环化预反应态选择性。值得注意的是,在500 ns轨迹中,开闭运动与预反应状态的形成和解散并不同步,蛋白质的动态运动导致共价结合的底物疏水尾端在口袋中翻转并形成环化预反应态。在环化预反应态中,其底物疏水尾端C11位的羟基与His259残基之间的氢键对形成环化预反应态发挥了引导作用,而Leu183、Leu186 和Leu190 非极性残基则形成起模板作业的疏水口袋,共同克服了酶内部共价结合底物疏水链尾端的大环构象变化带来的构象熵损失。预反应态计算也预测到Leu183、Leu186 和Val170 的丙氨酸突变可以改变酶催化环化和水解的倾向;其次,非极性氨基酸Phe260 与Ile79 突变为极性氨基酸则阻碍环化反应[95]。
硫酯酶对于6~8、12、14、16 元大环内酯的环合能力是药物生物合成的研究热点,为了进一步确认预反应态在硫酯酶选择催化水解或环化途径的重要性,我们比较了苦霉素硫酯酶催化环化12 元环(10-deoxymethynolide,甲霉素前体)和14 元环(narbonolide,苦霉素前体)中底物与硫酯酶共价中间体的动态行为[96]。与DEBS 硫酯酶相近,苦霉素硫酯酶也包含了活性口袋疏水界面、开放的底物通道和丝氨酸-组氨酸-天冬氨酸催化三联体,催化三联体位于通道的中心位置,底物与Ser148 形成共价中间体。对底物小分子体系开展构象搜索,从低能量构象中直接选取与催化口袋匹配的活性构象移植入酶中,以共价对接的方式构建了预反应态分子动力学模拟的出发结构,利用高斯软件构建丝氨酸-底物共价衍生结构RESP 电荷力场。利用短轨迹多次模拟的方法筛选出酶-底物共价中间体复合物的近进攻活性构象,然后对体系展开长轨迹模拟观察预反应态的结构稳定性。在各6 次50 ns 的短轨迹模拟中,有3 条12 元环和2 条14 元环轨迹中出现了较稳定His259-Asp169 氢键网络的环化反应活性构象。如果定义预反应态中氢键重原子距离小于0.30 nm、尾端羟基氧与丝氨酸连接的羧基碳原子之间的接触距离小于0.45 nm,则12 元环和14 元环体系在300 ns 轨迹中的His259 活性氢键总布居数分别为24.3%和14.2%,而12 元环碳氧短距离的活性构象比14 元环高出10.3%。尽管预反应态判定阈值因人为定义而几何结构参数具体数值的物理意义不甚清晰,从上述预反应态短轨迹模拟中可以观察到12 元环体系明显具有更多的环化反应活性构象,因此可以推导出12 元环的预反应态比14 元环占比更高、结构上更稳定。如果从这些短轨迹中选择最稳定的预反应态结构出发进一步把模拟时间推长到300 ns,12 元环和14 元环体系的氢键布居数分别为46.0%和27.5%,而短距离碳氧活性构象布居数分别为24.3%和14.2%,总布居数为11.2%和5.7%。从模拟轨迹中可以看到,除了类似红霉素硫酯酶预反应态分子模拟中组氨酸氢键引导作用之外,C9 位羧基与Thr77 侧链存在氢键也稳定预反应态结构,苦霉素硫酯酶在12 元环体系中疏水作用增加了Ala78 和Gly150残基。研究发现苦霉素硫酯酶的Tyr178 识别14 元环底物的作用不同于红霉素硫酯酶,前者为疏水相互作用,而后者中突变为疏水苯丙氨酸残基时失去催化活性。另一方面,预反应态的分子动力学模拟也展示了硫酯酶活性口袋的柔性,POVME口袋体积计算表明结合12 元环底物时口袋体积为0.335 nm3而14 元环底物时为0.355 nm3。QM/MM计算表明了后续的碳四面体结构形成和消除在反应势能面上14 元环的活化能皆高于12 元环。鉴于后续C-O 断裂和形成两步基元反应发生时除反应位点之外底物-酶复合物的拓扑几何结构变化很小,故可认为环化预反应态是由底物共价连接中间体决定。
变构霉素生物合成酶系硫酯酶则倾向于水解途径,产生线性产物分子,但如果在生物合成模块中调用苦霉素硫酯酶模块则生成了痕量14 元环合产物[97]。底物-酶共价中间体50 ns 短轨迹预反应态模拟结果表明,变构霉素硫酯酶体系中可以形成相当数量的组氨酸氢键(32.7%),与苦霉素硫酯酶结合变构霉素底物的体系中氢键布居数相近(33.5%);然而,变构霉素硫酯酶体系中短碳氧距离的活性构象相对较少(11.0%)而苦霉素硫酯酶体系中短碳氧距离的活性构象较高(22.7%),总布居数(9.3%)也低于苦霉素硫酯酶体系(13.6%)。如果采用相同的预反应态计算策略,300 ns长轨迹分析却给出相反的结果,变构霉素的环化预反应态更加稳定(保持高活性构象布居数,15.6%),而苦霉素的环化预反应态却持续稳定时间短(活性构象布居数减低到9.8%)。从环化预反应态动态形成过程来看,变构霉素的活性口袋中疏水和亲水性环境与苦霉素有较大差异,Try161形成了多重氢键提高了活性构象的稳定性。Y161F、Y161F&N179P、Y161A 的虚拟突变可有效改变短轨迹中环化预反应态的活性构象布居数(0.3%、12.0%和21.0%),说明了Tyr161 是决定环化预反应态布居数的关键残基。另外,变构霉素活性口袋中的水分子数目较大也导致了组氨酸残基活化水分子的水解活性构象大量增加,C1-Owater平均距离变短(变构霉素0.295 nm,苦霉素0.322 nm)、组氨酸与水分子之间氢键增强(变构霉素0.266 nm,苦霉素0.279 nm),水解预反应态活性构象布居数(23.1%)明显高于苦霉素体系(19.2%)。变构霉素体系研究表明,如果短轨迹与长轨迹的预反应态活性构象布居数出现反转现象,催化反应途径的竞争因素比较复杂,从传统分子对接方法推算预反应态与酶活可能会有比较大的偏差,需要进一步计算QM/MM反应势垒来确定多种反应途径之间的竞争关系[98]。
4.2 醇脱氢酶
药物生物合成中,不可避免会涉及高空间位阻的手性醇中间体合成,而烟酰胺腺苷二核苷酸依赖酶醇脱氢酶催化还原羧基化合物路径有非常重要的地位[99]。然而,天然来源的醇脱氢酶底物谱受限、立体选择性低、活性低,而且手性还原时受Prelog规则限制只能获得其中一种手性醇而不能获得其对映体[100]。虽然可通过定向进化方法等蛋白质工程技术提高其催化活性,受限于手性产物高通量筛选方法,目前半理性设计方法在立体选择性酶改造中有重要的价值。
克鲁维多孢酵母的醇脱氢酶可催化转化CPMK 手性合成药物中间体,我们对KpADH 酶立体选择性反转的两个高效突变体开展了预反应态分子动力学研究,以阐明决定立体选择性的因素[101]。该酶是一种典型短链脱氢酶,由NADPH提供氢负离子,而催化中心Tyr164 提供质子中和负氢原子进攻羧基而衍生出氧负离子洞。在催化口袋中Ser126 结合底物,Lys168 与Tyr164 形成稳定氢键网络极化酪氨酸酚羟基的质子而降低其pKa。早期的研究发现,非受约束的分子动力学模拟中底物无法保持在活性位点而扩散到溶剂中,通常在25 ns 内底物分子就离开反应中心,而酶的开合转换周期大约为20 ns[102]。因此,如果从传统的分子对接米氏复合物出发,长时间的分子模拟并不能有效获得预反应态布居数。为了提高计算效率,利用已知过渡态结构出发对预反应态进行多次重复短轨迹分子模拟,发现两种不同手性的预反应态结构布局在两个高效突变体中呈现完全不同的稳定性。Mu-R2 突变体的pro-R手性预反应态中,负氢原子和质子在非受约束的分子动力学模拟中可以稳定存在1 ns,而在pro-S手性预反应态中虽然负氢原子可以保持在范德华接触距离以内,但不能同时保持提供质子的Try164 氢键网络。而Mu-S5 突变体的pro-S手性预反应态中负氢原子和质子在非受约束分子动力学模拟中可以稳定存在1 ns 左右,在pro-R手性预反应态中虽然质子可以保持在氢键距离内,但负氢原子却远离羧基碳原子0.4 nm 以上。如果以氢键距离0.34 nm 和负氢原子距离0.45 nm 划分活性和非活性构象,Mu-R2的pro-R预反应态活性构象布居数(4.56%)是Mu-S5(0.52%)的8 倍左右,而Mu-S5 的pro-S预反应态活性构象布居数(7.64%) 是Mu-R2(1.84%)的4 倍以上。在Mu-R2 突变体中,可以看到Ala128、Phe161、Tyr127 和Phe197 与底物有很强的π-π 相互作用和疏水相互作用,而在Mu-S5中却没有,说明与硫酯酶类似,疏水作用在诱导契合活性构象中有非常重要的角色。另外,在氢键和负氢原子的距离分布图上,可以观察到布居数非常高的非活性构象,说明活性构象在分子动力学热平衡运动中并不占优势。类似变构霉素硫酯酶的环化预反应态形成机制,在短链醇脱氢酶的相对较小的活性口袋中二芳基酮底物的立体选择性反转也可能与预反应态的形成过程相关——与野生型对比,Mu-S5 的4 个突变位点N136、V161、C237 和G214 位于平行四边形的4 个角上,底物进入催化口袋时需要穿过这个“极门”而促使pro-S预反应态的形成,而底物上的氯取代刚好赋予极化苯环而吻合静电相互作用和π-π 相互作用。
在活性口袋空间相对较大的中链醇脱氢酶CpRCR 中,基于晶体结构的点突变分子模拟发现W286A 和F285A 可以扩大活性口袋的空间从而可以容纳更大的底物,而W116A 突变反而使得活性口袋空间塌缩导致显著的蛋白质构象重组。CpRCR 与4 个特定突变体具有高互补性,使得这些突变体可用于合成具有不同立体选择性的多种手性醇化合物。在CpRCR 中,由于羧基的两侧取代基中有一侧为小取代基,因此不同于二芳基酮底物,它可以在活性口袋内部翻转底物而改变预反应态的手性,立体选择性更多取决于两种不同手性预反应态的结合稳定性而非其形成过程[103]。
4.3 甲基转移酶DNMT3A
除了处于进化中的生物合成酶及需要人工超进化的工业酶之外,我们尝试了解预反应态在进化相对完善的催化元件中的地位。DNA 甲基转移酶3A(DNMT3A)是表观遗传甲基化的关键酶[104],在急性骨髓性白血病(AML)患者中高发R882H 突变[105],因此以正常型和致病型为研究对象可以了解进化后酶因残基突变干扰产生的预反应态结构变化[106]。首先,利用经典或加速分子动力学模拟产生甲基转移反应步的活性构象,以SAM辅因子中的硫原子与甲基碳原子之间的距离、甲基碳与靶标碱基C5 原子的距离搜寻在室温热运动可及的甲基转移活性构象,QM/MM 理论计算表明虽然突变可以同时稳定预反应态和过渡态,但反应势垒反而从正常型的14.9 kcal/mol升高到15.6 kcal/mol。对反应势垒的畸变-互作-势垒-张力(DIAS)能量分解分析表明,R882H 突变系统地降低了外围蛋白骨架能量(PRS 5 kcal/mol;TS 14.7 kcal/mol)。然而在反应中心部分,该突变一方面进一步提高TDI 的稳定性(4.9 kcal/mol),另一方面却减弱了TDTS 的稳定性(5.5 kcal/mol)。DNMT3A 酶的预反应态分析表明,对于进化比较完善的催化元件,反应势能面上的决速过渡态和中间体个数比较多,各个节点的控制度Xrate相近,虽然可以观察到远端突变通过准“牛顿摇篮”诱导契合机制对反应中心产生结构微扰。由于远端突变同时改变了过渡态和中间体,导致预反应态活性构象布居数与催化活性呈现非线性相关,单凭借分子动力学模拟提供的预反应态信息尚不足以确定分子进化已经相对完善的催化元件突变效应,需要系统地计算反应势能面内所有高控制度的TDTS和TDI。
4.4 磷酸吡哆醛依赖脱羧酶
磷酸吡哆醛(PLP,又称维生素B6)依赖酶具有多功能的特性,通常在休止状态时PLP 的醛基与酶中保守赖氨酸残基形成席夫碱中间体,底物氨基酸的氨基亲核进攻该中间体的亚氨基而置换出赖氨酸形成外醛亚胺(external aldimine)与PLP 交联。新形成的外醛亚胺中间体的碳-氮或碳-碳断裂分别发生转胺、消旋、β-消除、逆羟醛反应等多个候选反应途径[107]。我们从酪氨酸脱羧酶的晶体结构出发,构建外醛亚胺决速中间体(TDI),然后利用受约束的分子动力学模拟获得外醛亚胺脱羧反应的预反应态,通过对理论酶过渡态的内禀反应坐标展开前线轨道分析,发现预反应态与过渡态有非常相近的立体电子效应:负电荷从羧基转移到PLP的π 电子系统,碳-碳成键σ 轨道的电子流向C=N的π*轨道,这种超共轭效应是PLP 酶选择性催化碳碳键断裂的主要原因[108]。由此可见,碳碳键与碳氮双键保持相互垂直并且二面角为90°可以最大化σ-π*前线轨道的相互作用,这活性构象特征适用于PLP 依赖脱羧酶的各种突变体结合不同底物的脱羧反应。为了维系预反应态活性构象,His98、His241 必须利用氢键锚定即将离去的二氧化碳基团,使得碳氧键保持在一个特定的方向从而使得预反应态内反应前线轨道对齐,而Lys240-His241 的动态行为与PLP 进入活性位置相呼应,His241 的芳环与PLP 的芳环呈现π-π相互作用。预反应态分析与H241 的定点突变实验结果相符合,说明PLP 脱羧酶的外醛亚基的确是酶活控制度最大的核心预反应态。
4.5 N-氨甲酰-D-氨基酸酰胺水解酶
海因酶系是药物中间体D 型氨基酸的生物合成途径之一,其中包括了海因消旋酶、海因酶和N- 氨甲酰-D- 氨基酸酰胺水解酶 Dcarbamoylase[109]。在工业生产中,D-carbamoylase的热稳定性和活性都远逊于海因酶及消旋酶,我们对来自N.indicus的野生型及人工进化高效突变体M4 展开了预反应态分析,聚焦该酶催化反应中Cys171 亲核进攻底物酰胺的决速步[110]。在该预反应态中Lys126 稳定亲核进攻衍生出的氧负离子洞,而Glu47作为质子受体以盐桥稳定羧基,因为亲核进攻时亲核试剂的最高占据轨道需要对齐羧基的最低未占据π*轨道,亲核进攻的最适宜角度(Bürgi-Dunitz 角度)为107°左右。在野生型中,该角度分布在60°~100°或120°~150°,只有6.4%的构象在100°~110°之间。而在高效突变体M4 的预反应态中,亲核进攻角被优化到与Bürgi-Dunitz进攻角相匹配(26.4%),说明M4 的活性构象布居数提高了近4倍。进一步的结构分析发现,在野生型的活性口袋比较宽敞,而M4 高效突变体的口袋变窄使得底物与酶有更多的相互作用,Asn172 和Asn196 与底物形成了更稳定的氢键网络、Cys171与Arg174 的氢键作用倍增、His144 与Phe288/Pro198 之间的π-π 相互作用和疏水作用等有利因素。虽然这些相互作用的变化并不是直接来之M4高效突变体的4个位点(D187N、A200N、S207A、R211G),然而可以观察到M4 这些残基突变对活性口袋结合底物产生了明显的影响。
4.6 普鲁兰多糖酶
普鲁兰多糖酶是糖苷水解酶家族的重要成员之一,催化α-1,6-糖苷键水解反应,广泛应用于医药、材料、有机金属化学和糖化行业[111]。通过对同源酶的序列突变的交互熵分析,挖掘到协同进化残基对,进一步利用定点饱和突变扫描锁定位点,成功获得了远离活性中心2 nm 以上的高效突变体K631V/Q597K/D541I/D473E[112]。根据前人的QM/MM 计算我们对野生型和高效突变体展开了预反应态分子动力学模拟,该酶的决速步为催化中心天冬氨酸进攻α-1,6-糖苷键的SN2 取代反应,相应的预反应态活性构象的几何参数定义为Asp619的羧基与糖苷键C1 碳原子之间的距离和它们与离去基团烷氧基的夹角,计算结果表明碳氧距离的低四分位点在高效突变体中0.428 nm(最小值0.374 nm)而野生型为0.480 nm(最小值0.424 nm),进攻角的高四分位点在高效突变体中143°(最大值175°)而野生型为134°(最大值157°),说明普鲁兰多糖酶高效突变体的预反应态构象明显优于野生型。
4.7 冠状病毒主蛋白酶
冠状病毒的主蛋白酶(3C 蛋白酶)对于冠状病毒的生命活动极为重要,是抗冠状病毒药物设计的重要靶点。根据前人对SARS 病毒中主蛋白酶的QM/MM 的分子机制计算,催化口袋中His41 残基与Cys148 形成氢键,极化了Cys148 硫原子的亲核性[113]。因此,预反应态定义为包含Cys148 巯基负离子-His41 咪唑阳离子对的蛋白质复合物,通过对比MERS 冠状病毒野生型和M168L/T174V 突变型的预反应态分子动力学模拟中活性构象布局数,发现S-C 距离在二者之间并无太大区别(均可小于0.35 nm),说明Cys148巯基负离子都能够接近底物目标肽键碳原子,然而从亲核进攻角度而言,在突变型中巯基负离子能够更准确对准底物Gln-Ser 肽键的π*反键轨道——突变型进攻角中位数为86°而在野生型为82°,突变后与Bürgi-Dunitz 标准进攻角度107°更接近,因此它们的HOMO-LUMO 相互作用更强,进攻键角100°~110°二面角85°~90°范围的活性构象布居数之比为1∶1.8。鉴于分子动力学模拟中99%以上的构象都为休止状态(S-C 距离>0.48 nm),说明冠状病毒主蛋白酶与底物蛋白的进化尚未完全,M168L/T174V 突变仅部分提高了催化活性[114]。
在表1中对前期的预反应态研究做了一个系统的总结,所选择的催化元件反应势能面在前人的QM/MM 研究中皆已经报道,在预反应态模型分析中聚焦反应势能面控制点的选择、高效突变体或远端突变对催化中心几何结构和电子结构的微扰作用,以及活性构象布居数的变化。
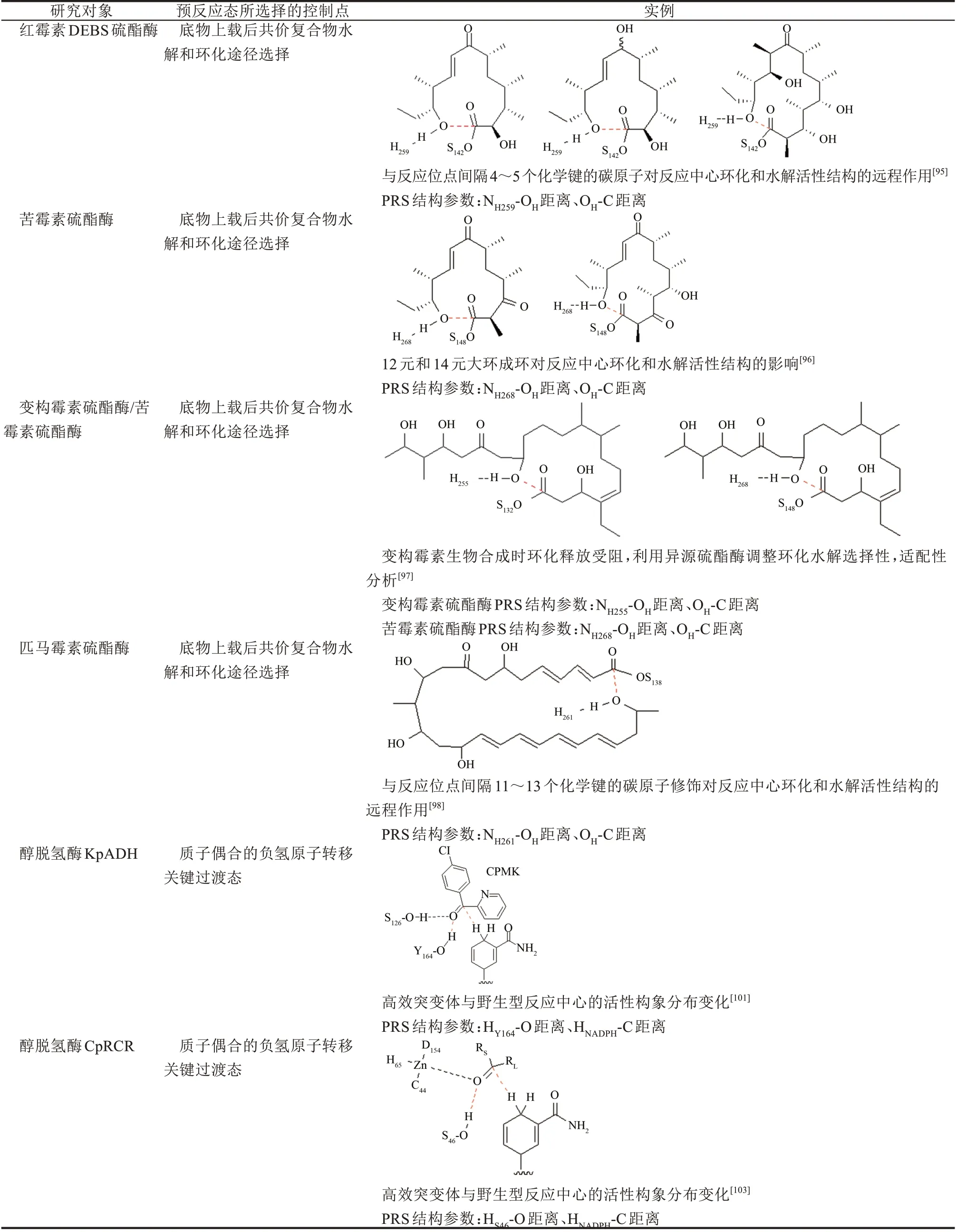
表1 预反应态模型的研究实例[95-98,101,103,106,108,110,112,114]Tab.1 Pre-reaction states of the studied enzymes,control points,and geometric parameters[95-98,101,103,106,108,110,112,114]
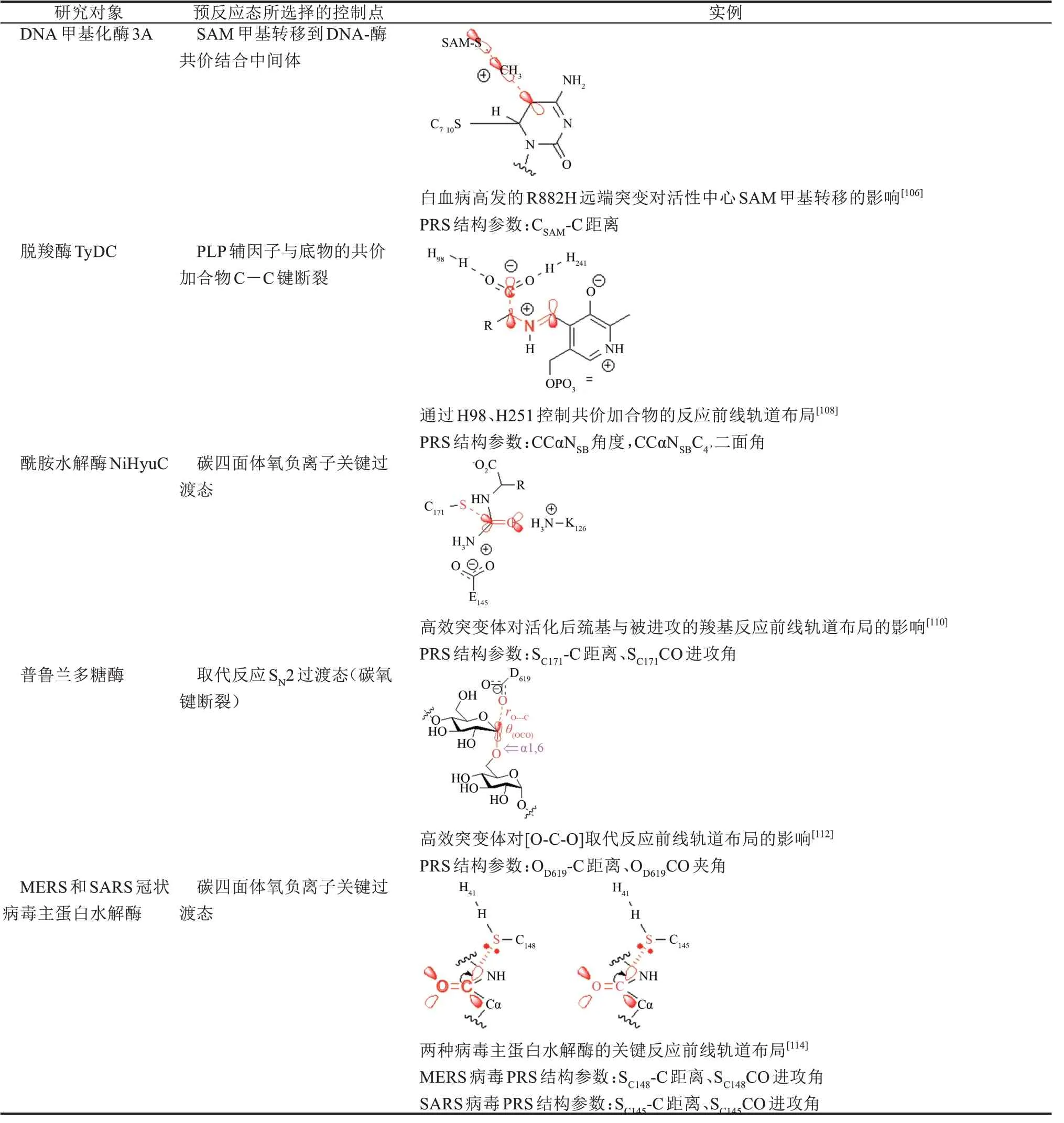
续表
5 挑战与展望
可见预反应模型基于催化元件的已知催化分子机制,假设蛋白质工程定向进化中催化循环反应途径并没有发生巨变,而是通过残基突变精修控制度较大的TDTS 和TDI 能量点,逐步达到人工进化完善状态,这一逐步优化逼近的过程与天然进化相似,由于在超级计算机中运行,其优化速度可以超过天然进化,有望达到超进化的合成生物学工程化要求。由于目前的通用型量子力学和分子动力学模拟的能量准确度尚不能达到实验精度(1 kcal/mol),关键步预反应态的选择上具有相当的随意性,通常是综合考虑分子进化对催化反应的化学键断裂和形成难易程度、多反应途径选择下的专一性(如手性选择性)、催化反应的可逆性等来确定需要优化的能量点,利用分子对接和手工搭建方法构建符合量子力学计算结果的活性构象,然后利用超级计算机GPU 分子动力学模拟的快速采样,分析催化元件氨基酸残基突变或周边环境对活性构象的影响。因此,预反应态模型可以用一个比较廉价的计算方案,在假设被拟合点对目标函数具有较高控制度的情况下,测算各种因素对实验结果的影响。预反应态计算可以覆盖催化元件外部环境对酶活性的影响,例如温度效应、酸碱效应、非水介质及小分子效应以及产物底物受限输运。通过对比不同介质(气相、水相、有机相、催化元件约束、Theozyme简化中心)的分子模拟,收集不同化学环境下预反应态的变化,提取和标记每个可以有效降低反应活化能的分子间相互作用(如极性氨基酸形成的氢键、离子对盐桥、离子-芳环相互作用等),可以建立催化中心保守性结构的化学信息,将一系列保守结构信息图谱归纳为“预反应态”的特征,从而可以利用信息学方法量化非酶催化反应转化为催化反应时机制变化,通过修改底物的化学结构和氨基酸残基及功能基团的空间布局,达到改变反应体系活性中心化学环境,优选出最佳反应通路。
预反应态不同于精细的量子力学计算,如基于inside-out 策略来改造或拓新催化反应机制,它更多地在对所获得的新酶分子初步定向进化后需要进一步改造的场景下利用已有海量实验酶活数据和已知催化分子机制,采用高性能计算建模来优化催化元件。预反应态模型结合突变文库酶活的大数据分析方法可以快速定位高效突变体对催化中心活性构象的影响,并进一步确定特定功能区和协同进化位点,确定nanozyme 和theozyme 为模板的酶催化反应的不同阶段的活性中心反应势能面、反应动力学和介质效应,验证了简化活性中心模型反应势能面上的关键中间体和过渡态在计算机辅助合理设计中的有效性和一致性,利用结构信息和能量信息分析活化焓和活化熵变化。利用实验数据-计算数据循环验证的方法自动化收集反应势能面上的关键点,为高阶量化过渡态计算提供反应坐标参数,避免在新酶计算机分子设计中人为搭建过渡态的盲目性。在预反应态计算中可以观察到催化动态结构对活性中心具有显著效应,与酶学中远端突变产生的动态效应相吻合,所用预反应模型具有内禀性的“dynamozyme”的概念,它来源于酶促反应分子机制中的活化熵变化,但底物分子受到催化元件约束时在原子运动水平上的动态特性。这种动态特性与催化元件的亚基多聚、嗜热或嗜冷、极端条件敏感性相关,将成为预反应态研究的热点。
