肌肉特异性表达Cas9 示踪同源打靶载体的构建及其在C2C12 细胞中的整合
2022-07-15王晓萌周慧敏董奕彤陈胜男安铁洙殷萍王春生
王晓萌,周慧敏,董奕彤,陈胜男,安铁洙,殷萍,王春生*
(1. 东北林业大学生命科学学院,哈尔滨 150040; 2. 黑龙江省医院,哈尔滨 150040)
外源基因的组织特异性表达在研究基因功能、细胞分化以及再生医学等领域具有广泛的应用前景。 组织特异性启动子是调控基因表达的“开关”,可实现外源基因在某些部位进行特异表达[1]。 骨骼肌具有生命周期长,操作方便,合成的蛋白容易进入体循环的特点[2-4],是基因治疗及动物模型构建的理想靶点,但肌肉特异载体的低表达水平限制了肌肉作为靶点的可能性。 因此,选择高转录效力的肌肉特异启动子尤为重要。 α-actin 是一种天然的骨骼肌启动子,可在骨骼肌细胞中特异表达,而在其他非肌肉细胞中几乎没有活性[5],但是由于受多种因素的影响,α-actin 的转录活性不稳定[6]。1999 年,Li 等[7]在对 α-actin 启动子和肌肉增强子的结构进行分析后,对肌肉特异性调控元件进行了重排与组装,合成包含 MEF-1、MEF-2、SRE 和TEF-1四种作用元件的肌肉特异启动子SP,其转录活性是α-actin 的 4 ~ 5 倍。 2021 年,Malerba 等[8]将 SP 启动子调控的抗肌萎缩蛋白MD1 腺病毒载体注射到肌萎缩小鼠模型中,SP 启动子驱动了MD1 在小鼠心肌中的高水平表达。
CRISPR/Cas 系统是细菌和古细菌抵御外来遗传物质的一种获得性免疫防御机制[9],包括能识别靶基因的CRISPR 序列和具有核酸酶活性的相关蛋白两个部分。 研究最为深入的CRISPR/Cas9 系统已被广泛运用于转基因动物的构建[10-13]。 在该系统中,sgRNA(single guide RNA)中存在一段与靶点PAM 序列(NGG)上游互补的序列,可引导Cas9 结合至靶位点,对靶DNA 的PAM 序列上游3 个碱基的位置进行切割使其双链断裂(DSB)[14]。 双链断裂的修复通过非同源末端连接(NHEJ)或同源定向修复(HDR)两种机制来实现。 因此,利用CRISPR/Cas9 系统,并提供外源基因同源载体作为HDR 的“原料”,即可实现外源基因的定点敲入。
利用CRISPR/Cas9 系统和组织特异启动子,可进行组织特异的基因编辑。 为构建肌肉特异表达Cas9 小鼠模型,本研究选取小鼠Rosa26 位点作为打靶位点,构建SP 启动子驱动的肌肉特异性表达Cas9 示踪同源打靶载体。 同时,这一研究也为构建疾病模型和基因疾病的相关治疗提供了新的方法。
1 材料与方法
1.1 材料
1.1.1 实验菌株,载体和细胞
感受态细胞DH5α(Cat# DL1001)购于上海唯地生物技术有限公司。 PX459-Rosa26 载体为实验室自存载体(载体上已连接 Rosa26 位点的sgRNA)。 Rosa26 供体载体购于广州易锦生物技术有限公司(Cat# DC-DON-SH02)。 载体详细信息见图1。 C2C12 细胞和293T 细胞为实验室前期冻存细胞。
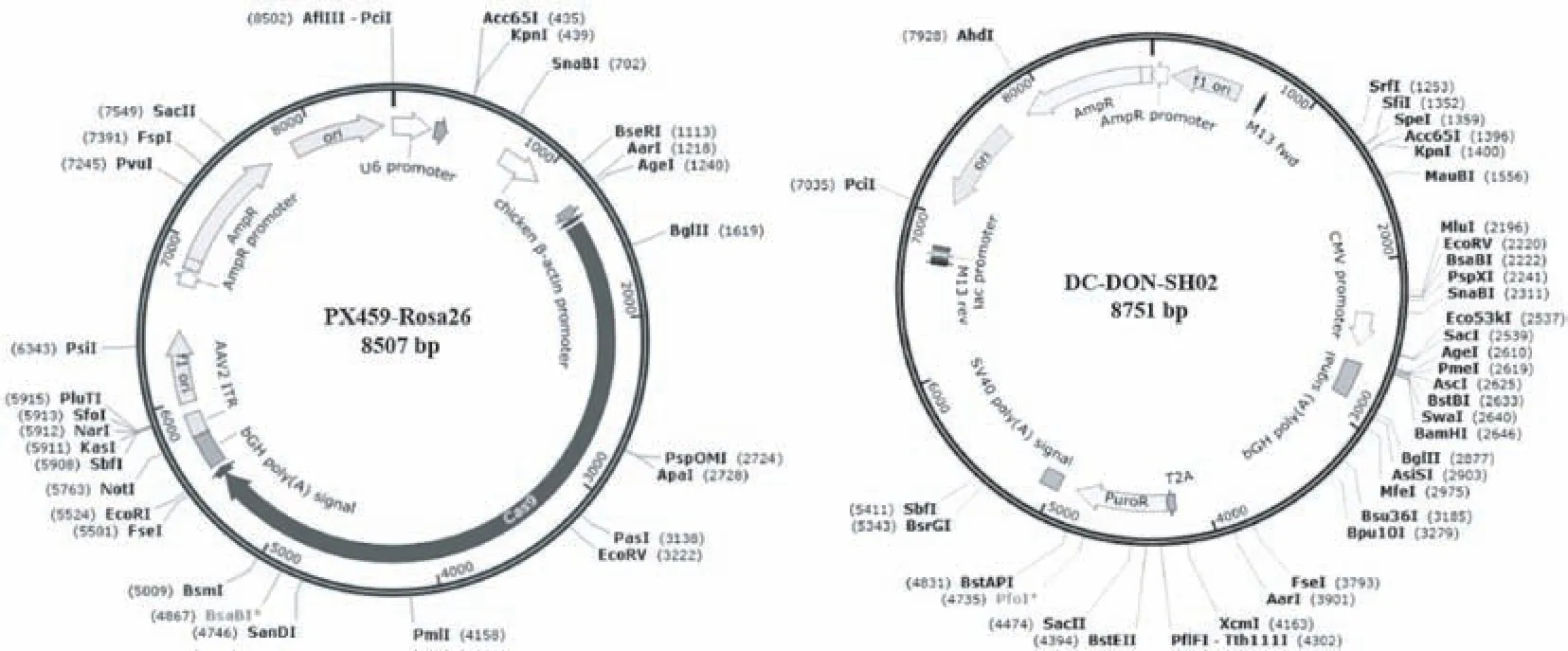
图1 载体示意图Figure 1 Diagram of plasimd
1.1.2 主要试剂与仪器
2×Vazyme LAmp Master Mix(Vazyme,P312-01),2 × ES Taq Master Mix(CWBIO,CW0690),Endo-free Plasmid Mini Kit II(Omega,D6950-01),Gel Extraction Kit (CWBIO,CW2302M),Lipfectamine 2000(Invitrogen,11668030),GeneJET Genomic DNA Purification Kit (Thermo, K0721), T4 DNA Ligase(TAKARA,2011A),T7 Endonuclease I 酶(NEB,E3321),同源重组酶(Vazyme,C112-01),限制性内切 酶KpnI ( Promega, R6341),AgeI ( Promega,R7251),XbaI(Promega,R6181),EcoRI(Promega,R6011),MluI (Promega, R6381),BstBI (Thermo,ER0121)。
PCR 仪(Applied Biosystems,9700,美国),分光光度计(Thermo,NanoDrop 2000,美国),倒置荧光显微镜(Carl Zeiss,Axiovert 200,德国),CCD 成像系统(Nikon,DS-Fi1,日本),离心机(Beckman Coulter,Microfuge 16,美国)。
1.2 方法
1.2.1 肌肉特异性表达Cas9 载体的构建
(1)SP 启动子的合成与PCR 扩增:SP 序列参照Li 等[7]的报道,由南京金斯瑞生物科技公司合成后连接于 pUC57 载体,测序正确的载体命名为pUC57-SP。 利用Primer 5 软件设计SP 启动子上下游引物并命名为SP-F 和SP-R(引物序列见表1),在上下游引物的5’端分别引入KpnI 和AgeI 的酶切位点,引物交由哈尔滨睿博兴科生物技术有限公司合成(本研究所用引物均由该公司合成)。 以载体pUC57-SP 为模板,进行 SP 启动子的 PCR 扩增。 反应组成:模板 10 ng,SP-F 1 μL,SP-R 1 μL,2 × ES Taq Master Mix 12.5 μL,补加 ddH2O 至 25 μL。PCR 反应程序:95℃ 5 min(预变性);95℃ 30 s(变性),62℃ 30 s(退火),72℃ 90 s(延伸),共 31 个循环;72℃ 7 min(终延伸)。 1%琼脂糖凝胶电泳检测后对目的条带进行胶回收。
(2) PX459-Rosa26-SP 的连接:PX459-Rosa26为连有打靶Rosa26 位点sgRNA 的 PX459 载体。 采用KpnI 和AgeI 分别对 SP 启动子扩增片段与PX459-Rosa26 载体双酶切,电泳检测后对目的条带进行胶回收。 将上述获得的载体长片段和SP 启动子进行连接,反应组成:SP 回收产物40 ng,PX459-Rosa26 回收产物 100 ng,T4 DNA 连接酶 0.5 μL,10 × Buffer 1 μL,补加 ddH2O 至 10 μL。 连接条件为4℃ 16 h。 连接产物使用感受态细胞DH5α 进行转化,挑取单个菌落进行培养,按照质粒小提试剂盒说明书对菌液进行提取,所得质粒经KpnI 和AgeI酶切鉴定后送至哈尔滨睿博兴科生物技术有限公司测序(本研究所有测序均由该公司完成)。
1.2.2 PX459-Rosa26-SP 在C2C12 细胞中编辑活性检测
(1)细胞转染与DNA 的提取:C2C12 细胞培养条件为37℃、5% CO2饱和湿度。 传代后,待细胞融合度达到60% ~70%,使用Lipfectamine 2000 转染试剂转染PX459-Rosa26 或PX459-Rosa26-SP 载体,未转染的C2C12 细胞作为对照组。 转染72 h 后,提取C2C12 细胞 DNA。
(2)打靶位点的 PCR 扩增:Rosa26 序列中,XbaI 酶切位点为PX459-Rosa26-SP 打靶位点,利用Primer 5 软件在XbaI 酶切位点左右为207 bp 和398 bp 处设计引物并命名为 mRosa26-XbaI-F1 和mRosa26-XbaI-R1(引物序列见表1),扩增产物理论值为 600 bp。 对 C2C12 DNA 进行 PCR 扩增,电泳检测后对目的条带进行胶回收。
(3)编辑效率检测:采用两种方法检测SP 启动子和CMV 启动子带动 Cas9 蛋白的编辑效率。 对PCR 产物进行限制性内切酶XbaI 酶切检测。 反应组成:XbaI 0.5 μL,PCR 胶回收产物 200 ng,10 × FD Buffer 1 μL,补加 ddH2O 至 10 μL。 酶切条件为 37℃1 h。 2%的琼脂糖凝胶电泳检测酶切情况。
对PCR 产物进行 T7E1 酶切检测。 PCR 产物退火 杂 交, 反 应 组 成: PCR 产 物 200 ng, 10 ×NEBuffer2 2 μL,补加 Nuclease-free Water 至 19 μL。退火杂交条件:95℃ 5 min(预变性);95 ~85℃,-2℃ /s;85 ~ 25℃,-0.1℃ /s(条件降温)。 杂交后的产物经T7E1 酶切鉴定。 反应组成:退火反应产物 19 μL,T7E1 1 μL。 酶切条件为 37℃水浴 15 min后加入 0.25 mol/L 的 EDTA 1.5 μL 终止反应。 2%的琼脂糖凝胶电泳检测酶切情况。
1.2.3 PX459-Rosa26-SP-DsRed 示踪载体的构建
(1)T2A-DsRed 片段的PCR 扩增:根据实验室保存的含有T2A-DsRed 的载体序列,利用Primer 5软件设计T2A-DsRed 上下游引物并命名为T2ADsRed-F 和 T2A-DsRed-R,引物 5’端引入同源臂(引物序列见表 1)。 通过 PCR 进行 T2A-DsRed 的扩增。 反应组成:T2A-DsRed-F 1 μL,T2A-DsRed-R 1 μL,模板10 ng,2 × ES Taq Master Mix 12.5 μL,补加 ddH2O 至 25 μL。 反应程序:95℃ 5 min(预变性);95℃ 30 s(变性),62℃ 30 s(退火),72℃ 45 s(延伸),共 32 个循环;72℃ 7 min(终延伸)。 电泳检测后对目的条带进行胶回收。
(2)PX459-Rosa26-SP-DsRed 的连接:将得到的T2A-DsRed 片段与经EcoRI 酶切后的 PX459-Rosa26-SP 载体进行同源重组。 反应组成:T2ADsRed 片段 20 ng,PX459-Rosa26-SP 载体大片段 80 ng,Exnase Ⅱ 1 μL,5 × CE Ⅱ Buffer 2 μL,补加ddH2O 至 10 μL。 同源重组条件为 37℃ 30 min,立即至于冰上冷却。 同源重组产物使用感受态细胞DH5α 进行转化,提取质粒后测序。
1.2.4 PX459-Rosa26-SP-DsRed 示踪载体的检测
按1.2.2 中的条件和方法对C2C12 细胞进行培养和PX459-Rosa26-SP-DsRed 载体的转染,设置未转染的C2C12 细胞作为对照组。 48 h 后,观察荧光表达情况。
1.2.5 肌肉特异表达Cas9 同源打靶示踪载体的构建
(1)SP-Cas9-DsRed 的扩增:利用 Primer 5 软件根据PX459-Rosa26-SP-DsRed 载体SP 启动子的上游和DsRed 下游设计引物,将 Donor 载体MluI 与BstBI 酶切位点对应的同源臂分别引入上下游引物,并命名为T-MluI-SP-F 和T-BstBI-R(引物序列见表1)。 以 PX459-Rosa26-SP-DsRed 为模板,PCR 扩增。 反应组成:T-MluI-SP-F 1 μL,T-BstBI-R 1 μL,载体 10 ng,2×Vazyme LAmp Master Mix 12.5 μL,补加 ddH2O 至 25 μL。 PCR 反应程序:98℃ 2 min(预变性);98℃ 30 s(变性),65℃ 30 s(退火),72℃5 min(延伸),共 30 个循环;72℃ 7 min(终延伸)。电泳检测后对目的条带进行胶回收。
(2)同源臂载体 Donor 的酶切:利用MluI 和BstBI 对DC-DON-SH02 Rosa26 供体载体进行大量双酶切,电泳检测后对大片段进行胶回收。
(3)Donor-Cas9-SP-DsRed 示踪载体的连接:将得到的SP-Cas9-DsRed 片段与Donor 载体大片段进行同源重组。 反应组成:SP-Cas9-DsRed 片段 40 ng,Donor 载体大片段 50 ng,Exnase Ⅱ 1 μL,5 × CEⅡ Buffer 2 μL,补加 ddH2O 至 10 μL。 同源重组条件为37℃ 30 min,立即至于冰上冷却。 同源重组产物使用感受态细胞DH5α 进行转化,提取质粒,所得质粒经MluI 和BstBI 酶切鉴定后测序。
1.2.6 Donor-Cas9-SP-DsRed 示踪载体在 C2C12 细胞中整合情况的检测
(1)细胞转染及荧光观察:按1.2.2 中的条件和方法对C2C12 细胞进行培养以及PX459-Rosa26载体与 Donor-SP-Cas9-DsRed 的共转染。 48 h 后,对荧光表达情况进行观察。
(2)同源打靶载体整合情况的PCR 鉴定:转染48 h 后加入 Puro 筛选 7 d,提取细胞 DNA。 利用Primer 5 软件设计两对鉴定引物并命名为 5’-Rosa26-F,5’-SP-R 和 3’-Poly(A)-F,3’-Poly(A)-R(引物序列见表1)。 对C2C12 细胞DNA 进行 PCR扩增。 反应组成:上下游引物各 1 μL,2×Vazyme LAmp Master Mix 12.5 μL,C2C12 细胞 DNA 10 ng,补加 ddH2O 至 25 μL。 PCR 反应程序:98℃ 2 min(预变性);98℃ 30 s(变性),62℃ 30 s(退火),72℃5 min(终延伸),共 30 个循环;72℃ 7 min(终延伸)。 电泳检测后对目的条带进行胶回收和测序。

表1 引物序列Table 1 Primer sequences
2 结果
2.1 肌肉特异性表达Cas9 载体PX459-Rosa26-SP的构建
PX459-Rosa26 为实验室前期构建的载体,该载体上已连有打靶 Rosa26 位点 sgRNA,因此,选用PX459-Rosa26 作为骨架进行改造(图2A)。 使用含有限制性内切酶KpnI 和AgeI 酶切位点的引物SP-F和SP-R 对PUC57-SP 载体进行SP 启动子的扩增。电泳结果显示,在约300 bp 处观察到与理论值相符的特异性条带(图2B),即SP 启动子扩增成功。 用限制性内切酶KpnI 和AgeI 分别对SP 启动子片段和载体PX459-Rosa26 进行酶切,如图2C 所示,在约800 bp 处出现PX459-Rosa26 载体小片段,回收SP启动子目的片段及载体大片段并进行连接。 对重组质粒进行KpnI 和AgeI 双酶切鉴定,结果显示,在约7800 bp 和300 bp 处出现目的条带(图2D),与理论值相符。 重组质粒进行测序鉴定,目的片段未发生突变,该重组质粒命名为PX459-Rosa26-SP。
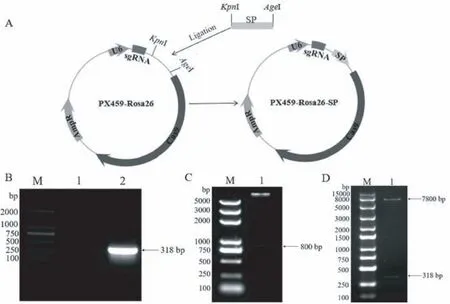
图2 PX459-Rosa26-SP 载体的构建Note. A. The diagram of PX459-Rosa26-SP construction. B. Results of agarose gel electrophoresis of SP promoter PCR product. M. DL-2000 Marker. 1. Control. 2. PCR product. C. Results of agarose gel electrophoresis of SP promoter enzyme digestion product. M. DL-5000 Marker. 1.Enzyme digestion product. D. Results of agarose gel electrophoresis of PX459-Rosa26-SP enzyme digestion product. M. 250 bp DNA Ladder. 1.Enzyme digestion product.Figure 2 Construction of PX459-Rosa26-SP vector
2.2 肌肉特异性表达Cas9 载体PX459-Rosa26-SP编辑效率的检测
将 pAcGFP1-N1 载体,PX459-Rosa26 载体,PX459-Rosa26-SP 载体分别转染C2C12 细胞,同时设置一组未转染载体的C2C12 细胞为阴性对照。 48 h 后,转染了GFP 载体的C2C12 细胞观察到绿色荧光表达,表明外源质粒已经转染至C2C12 细胞。 转染72 h 后,分别提取转染 PX459-Rosa26 载体,PX459-Rosa26-SP 载体和未转染的3 组细胞的DNA。 使用引物mRosa26-XbaIF1 和mRosa26-XbaI-R1 分别对3 组细胞的DNA 进行扩增。 如图3A 所示,在约600 bp 处出现目的条带。PCR 产物进行XbaI 酶切后,理论上会被切为200 bp 与400 bp 两个片段。 酶切产物进行电泳后,结果显示,未转染的细胞Rosa26 位点被完全切开,转染PX459-Rosa26 载体和PX459-Rosa26-SP 载体的两组Rosa26 位点均未被完全切开(图3B),这是XbaI 酶切位点发生突变后使酶切效率降低导致的。 使用T7E1 酶对载体编辑效率进行进一步检测,电泳结果显示(图3C),未转染载体的对照组未发生切割,转染PX459-Rosa26 载体和PX459-Rosa26-SP 载体的酶切不完全。 实验结果使用Image Lab 软件分析。 灰度分析显示,转染PX459-Rosa26 载体的突变概率为46.42%,转染载体PX459-Rosa26-SP 的突变概率为18.38%。

图3 PX459-Rosa26-SP 编辑效率检测Note. A. Results of agarose gel electrophoresis of C2C12 cell DNA PCR products. M. DL-2000 Marker. 1. PCR product of cells transfected with PX459-Rosa26-SP. 2. PCR product of cells transfected with PX459-Rosa26. 3. PCR product of cells without transfected. B. Results of agarose gel electrophoresis of XbaⅠdigestion product. M. DL-2000 Marker 1. Digestion product of cells transfected with PX459-Rosa26-SP. 2. Digestion product of cells without transfected. 3. Digestion product of cells transfected with PX459-Rosa26. C. Results of agarose gel electrophoresis of T7E1 digestion product. M. DL-2000 Marker. 1. Digestion product of cells without transfected. 2. Digestion product of cells transfected withPX459-Rosa26-SP. 3. Digestion product of cells transfected withPX459-Rosa26.Figure 3 Editing efficiency detection of PX459-Rosa26-SP
2.3 肌肉特异性表达 Cas9 示踪载体 PX459-Rosa26-SP-DsRed 的构建及表达活性检测
PX459-Rosa26-SP-DsRed 载体构建过程见图4A。 以实验室保存的连有T2A-DsRed 的载体为模板,使用含有限制性内切酶EcoRI 酶切位点的引物T2A-DsRed-F 和 T2A-DsRed-R 进行 T2A-DsRed 的扩增。 PCR 产物的电泳结果显示,在约740 bp 左右处观察到与理论值相符的特异性条带(图4B),即T2A-DsRed 扩增成功。 用限制性内切酶EcoRI 对载体 PX459-Rosa26-SP 进行酶切,在800 bp 左右处出现小片段(图4C)。 回收T2A-DsRed 片段及PX459-Rosa26-SP 载体大片段进行同源重组,对重组质粒进行测序鉴定鉴定,目的片段未发生突变,该重组质粒命名为 PX459-Rosa26-SP-DsRed。 将 PX459-Rosa26-SP-DsRed 载体转染至 C2C12 细胞中(图4D),转染48 h 后,利用绿光照射C2C12 细胞,可以观察到红色荧光(图4E),表明SP 启动子可带动红色荧光蛋白基因在成肌细胞中表达。

图4 PX459-Rosa26-SP-DsRed 构建及表达活性分析Note. A. The diagram of PX459-Rosa26-SP-DsRed construction. B. Results of agarose gel electrophoresis of T2A-DsRed PCR product. M. DL-2000 Marker. 1. PCR product. C. Results of agarose gel electrophoresis of PX459-Rosa26-SP digestion product. M. Trans8K DNA Marker Marker.1. Enzyme digestion product. D. C2C12 cells transfected with PX459-Rosa26-SP-DsRed (Brightfield). E. C2C12 cells transfected with PX459-Rosa26-SP-DsRed (Excitation field).Figure 4 Construction and activity analysis of PX459-Rosa26-SP-DsRed
2.4 肌肉特异表达 Cas9 示踪同源打靶载体Donor-SP-Cas9-DsRed 的构建及检测
Donor-SP-Cas9-DsRed 载体构建过程见图5A。以PX459-Rosa26-SP-DsRed 为模板,扩增 SP-Cas9-DsRed 片段,PCR 产物经电泳检测,如图5B 所示,在约5300 bp 处观察到与理论值相符的特异性条带,表明SP-Cas9-DsRed 扩增成功。 用限制性内切酶MluI 和BstBI 对Donor 载体进行双酶切,电泳结果显示,在8300 bp 和500 bp 左右处出现两条条带,与理论值相符(图5C)。 回收 SP-Cas9-DsRed 片段及Donor 载体大片段进行同源重组,对重组质粒进行酶切鉴定,如图5D 所示,在约8300 bp 和5400 bp处出现目的条带,与理论值相符。 测序结果显示,目的片段未发生突变,该重组质粒命名为Donor-SPCas9-DsRed。 将 Donor-SP-Cas9-DsRed 载体转染至C2C12 细胞中(图5E),同时转染293T 细胞作为对照组。 48 h 后,C2C12 细胞中观察到红色荧光与绿色荧光(图5F,5G),而对照组的293T 细胞只观察到绿色荧光的表达(图略),表明所构建的Donor-SP-Cas9-DsRed 载体能在细胞中正常表达且具有细胞特异性。
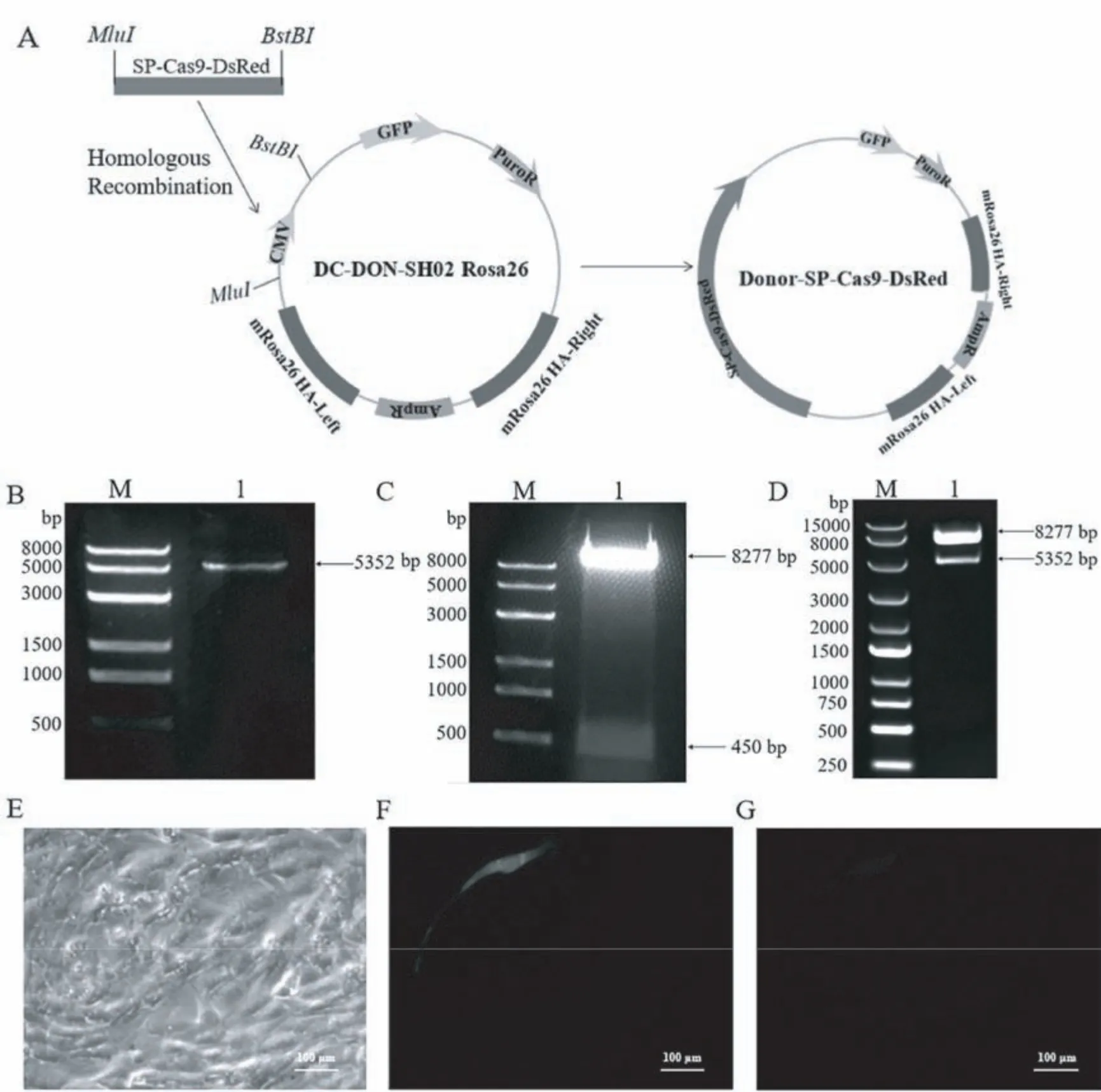
图5 Donor-SP-Cas9-DsRed 构建及表达活性分析Note. A. The diagram of Donor-SP-Cas9-DsRed construction. B. Results of agarose gel electrophoresis of SP-Cas9-DsRed PCR product. M.Trans8K DNA Marker. 1. PCR product.C. Results of agarose gel electrophoresis of Donor digestion product. M. Trans8K DNA Marker. 1. Enzyme digestion product. D. Results of agarose gel electrophoresis of Donor-SP-Cas9-DsRed enzyme digestion product. M. 250 bp DNA Ladder. 1. Enzyme digestion product. E. C2C12 cells transfected with Donor-SP-Cas9-DsRed (Brightfield). F. C2C12 cells transfected with Donor-SP-Cas9-DsRed(C2C12 cell expressed GFP). G. C2C12 cells transfected with Donor-SP-Cas9-DsRed (C2C12 cell expressed DsRed).Figure 5 Construction and activity analysis of Donor-SP-Cas9-DsRed
2.5 肌肉特异表达 Cas9 示踪同源打靶载体Donor-SP-Cas9-DsRed 在C2C12 细胞中整合
将PX459-Rosa26 载体和Donor-SP-Cas9-DsRed载体共转染至 C2C12 细胞中(图 6A),48 h 后,C2C12 细胞表达绿色荧光和红色荧光(图6B,6C)。对转染后的细胞进行终浓度为1.1 μg/μL 的Puro筛选。 7 d 后,细胞表达绿色荧光和红色荧光,提取细胞 DNA 进行 PCR 鉴定。 图 6D 为 3’ 端插入情况,在约1200 bp 处出现特异性条带,与理论值相符。 图 6E 为5’端插入情况,在约 1000 bp 处出现与理论值相符的特异性条带,PCR 产物回收后进行测序,测序结果与理论序列相符,表明构建的Donor-SP-Cas9-DsRed 载体整合到 C2C12 细胞 Rosa26位点。

图6 Donor-SP-Cas9-DsRed 整合检测Note. A. C2C12 cells after co-transfection (Brightfield). B. C2C12 cells after co-transfection (C2C12 cell expressed GFP). C. C2C12 cells after co-transfection (C2C12 cell expressed DsRed). D. Results of agarose gel electrophoresis of 3’DNA PCR product. M. DL-2000 Marker. 1. PCR product. E. Results of agarose gel electrophoresis of 5’DNA PCR product. M. DL-2000 Marker. 1. PCR product.Figure 6 Cell intergation detection of Donor-SP-Cas9-DsRed
3 讨论
与“锌指核酸内切酶(ZFN)”和“类转录激活因子效应物核酸酶(TALEN)”技术相比[15-16],第三代“基因组定点编辑技术”CRISPR/Cas9,具有成本低、制作便捷以及快捷高效等优点,并已成为科学研究领域的有效工具[17-18]。Rosa26 位点位于小鼠六号染色体上,被应用于整合转基因构建体以在小鼠中实现普遍存在或条件基因表达[19]。Rosa26 由3 个外显子构成,其转录产物为一种非必需、非编码的RNA,并且不会翻译成蛋白质[20]。 因此,Rosa26 位点也有“安全港”之称。 经研究证实,Rosa26 基因在大部分组织和细胞中都有表达,因此,在此区域定点插入外源DNA,在各组织中表达的可能性都会非常高[21]。Rosa26 位点基因编辑技术可以非常有效地建立多用途的条件性转基因动物模型,因此,本研究选取Rosa26 位点作为打靶位点进行研究。
对特定组织进行基因改造时,需要保证个体其它组织器官正常的生长发育不受影响。 为实现肌肉特异打靶的同时不损害其它组织器官的生理功能,本研究选用肌肉特异启动子SP 驱动Cas9 的表达,并以打靶小鼠Rosa26 位点的Cas9 载体PX459-Rosa26 为骨架,构建肌肉特异表达 Cas9 载体PX459-Rosa26-SP。 C2C12 细胞作为骨骼肌前体细胞,是研究成肌细胞增殖分化的理想模型[22]。 随后,我们对成肌细胞C2C12 进行转染和基因组编辑效率的检测。 转染PX459-Rosa26-SP 后,提取细胞基因组并进行酶切验证,突变的概率为18.38%,表明SP 启动子可带动Cas9 蛋白在C2C12 细胞中表达。 之后,我们在载体Cas9 后引入DsRed 红色荧光报告基因,可通过荧光的表达来直观的观察SP 启动子在不同细胞中的特异表达情况。 在去除Rosa26 位点 Donor 载体 ORF 区的 CMV 启动子后,我们在该载体骨架上连接SP-Cas9-DsRed,构建肌肉特异表达Cas9 示踪同源打靶载体Donor-SP-Cas9-DsRed。 将 Donor-SP-Cas9-DsRed 和 PX459-Rosa26共转至C2C12 细胞后,细胞可表达绿色荧光和红色荧光,且这种现象是肌肉细胞特异性的,即SP 启动子可启动外源基因的肌肉特异性表达。 对转染后的C2C12 细胞进行Puro 筛选后得到阳性细胞进行基因组的提取和 PCR 鉴定,该载体成功整合到C2C12 细胞Rosa26 位点。 我们构建的系统可用于肌肉特异表达Cas9 小鼠模型的制备和肌肉特异性基因编辑的研究。
