二例遗传性大疱性表皮松解症基因突变研究
2022-07-15廖晓捷于越乾王真真孙乐乐张福仁
廖晓捷 于越乾 王真真 孙乐乐 刘 红 张福仁
山东第一医科大学附属皮肤病医院(山东省皮肤病医院),山东省皮肤病性病防治研究所,山东济南,250022
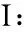
遗传性大疱性表皮松解症(epidermolysis bullosa,EB)是一种罕见且严重的机械性皮肤病,在新生儿中的发病率约为19/106、患病率约为11/106,患者以皮肤或黏膜脆性增加、摩擦或创伤后水疱、大疱的形成为主要临床特征[1]。我们对2例EB患者进行调查,应用全外显子组测序技术进行分析,再用Sanger测序验证结果,为临床诊断提供确切的依据。
1 资料与方法
1.1 临床资料 山东第一医科大学附属皮肤病医院门诊收集2例EB患者。散发患者1:女,19岁。四肢出现淡粉红色或褐色的丘疹、结节3年,伴剧烈瘙痒,表面破溃,部分皮损愈合后可见结痂和萎缩性瘢痕。患者无类似疾病家族史,其父母非近亲结婚。皮肤科检查:双前臂、双胫前和双足背可见大小不等的淡粉红色或褐色的丘疹、结节和结痂,未见明显水疱,无血疱(图1a);双足第一趾甲增厚萎缩(图1b),口腔黏膜未受累。皮肤组织病理示:表皮角化过度、灶性角化不全,表皮下裂隙,真皮浅层血管、纤维组织增生,血管周围轻度单一核细胞浸润,符合大疱性表皮松解症。结合患者的临床表现和皮肤组织病理检查,临床诊断为EB。
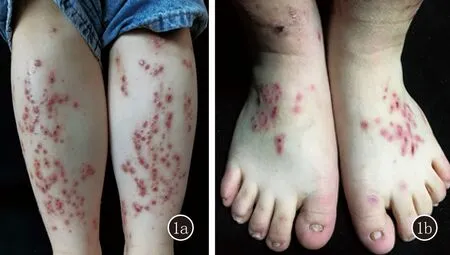
图1 散发患者1临床照片 双胫前紫红色结节、丘疹,部分结痂(1a),双足第一趾甲增厚萎缩、营养不良(1b)
家系患者2:先证者,男,1岁。自出生双手足背出现白色粟丘疹、水疱,伴右手中指指甲脱落(图2a)。先证者足月生产,一般情况好,父母非近亲婚配。皮肤科检查:双手足背白色粟丘疹、水疱伴皮肤破损,右手中指指甲脱落,无自觉症状,无血疱,手掌、肘部、膝部等摩擦部位和口腔黏膜均未受累。其父有类似病史,自幼双手足背水疱,摩擦后加重,随年龄增长水疱逐渐减轻,但出现双胫前粟丘疹,伴剧烈瘙痒,可见糜烂结痂,愈合后留有白色萎缩性瘢痕;双手足指趾甲均变薄、浑浊,远端缺损(图2b),未曾出现过手掌、足趾等部位的紧张性大疱。结合病史及皮损表现,临床拟诊为EB。先证者幼小,其父母拒绝对其行皮肤组织病理检查和抽血检查,为进一步明确诊断,对患儿父亲进行全基因组外显子测序。
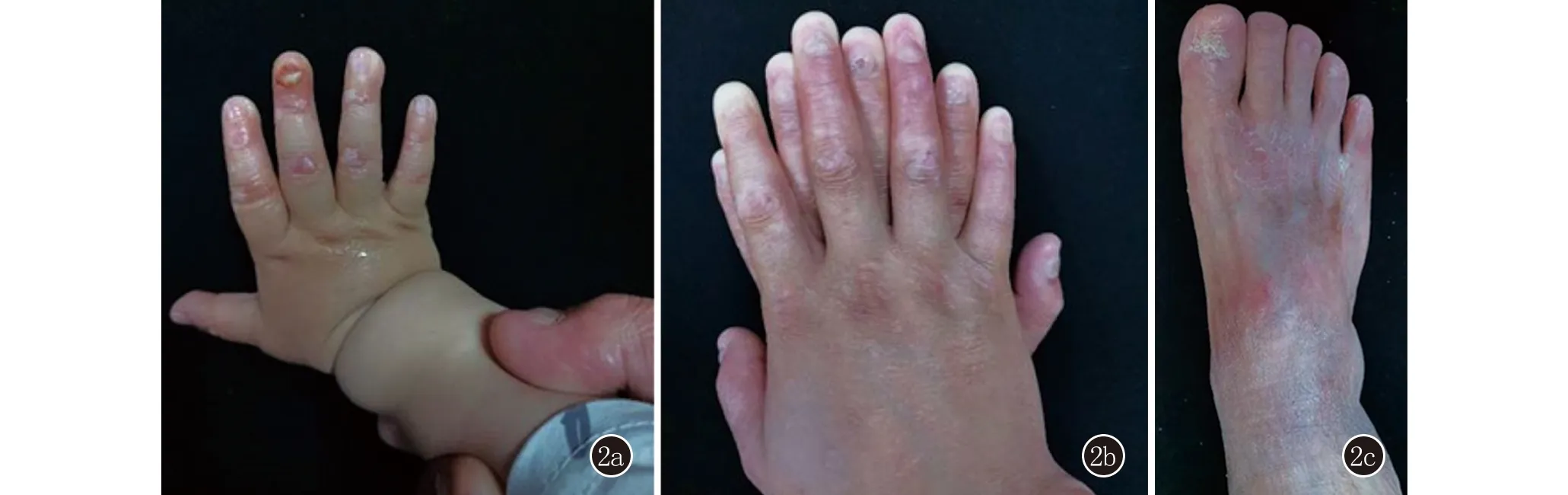
2a:先证者双手白色粟丘疹、水疱,伴右手中指指甲脱落;2b,2c:其父双手足白色粟丘疹,双手足指趾甲均变薄、浑浊,远端缺损
1.2 研究方法
1.2.1 样本采集 经本院伦理委员会批准,经过家系成员的知情同意并签字后,采集患者及其父母的外周血5 mL,EDTA抗凝,采用全自动核酸提取仪提取所有标本DNA,使用核酸浓度测定仪NanoDrop2000对标本 DNA的含量及纯度进行测定,并将其标化为30 ng/μL的DNA模板。
1.2.2 变异筛选 将散发患者1、家系2先证者父亲的外周血 DNA进行全基因组外显子测序,首先使用AIExome Enrichment Kit V4试剂盒进行捕获实验和构建文库,基因组文库经过质量检测合格后通过 Illumina HiSeq 6000 平台完成高通量测序。测序原始数据采用BWA 软件与参考基因组(hg19)进行比对,比对好的数据用GATK软件进行变异位点的寻找,寻找的位点均通过Annovar软件评估变异的影响并进行注释。
1.2.3 Sanger测序验证 根据全基因组外显子测序筛选出的致病基因突变位点,通过NCBI数据库查取突变基因的外显子序列,应用Primer5.0软件设计引物,对收集的外周血DNA样本进行突变位点的Sanger测序验证。PCR反应条件: 预变性94℃ 10 min、变形 94℃ 30 s、退火55℃ 45 s、延伸 72℃ 10 min,共35个循环之后72℃延伸10 min;取PCR反应产物经琼脂糖凝胶电泳,将目的片段切胶纯化后用ABI 3500Xl Dx Genetic Analyzer测序仪进行检测。
1.2.4 突变分析 所有测序结果经NCBI blast数据库、Chromas软件与相应的突变基因参考序列进行分析比对,检索Pubmed和万方数据库至今发表的关于本研究中突变位点的文章。
2 结果
2.1 临床调查 调查患者的临床资料,其中类似患者2例,分别为散发患者1和家系2先证者的父亲,临床症状均相似:均表现为双胫前痒疹样改变,伴有轻度甲营养不良;但发病年龄不同,前者是近成年才出现皮损,后者是出生后即有皮损,且水疱发生于四肢伸侧,尤其是趾指关节面,愈后留有白色丘疹状瘢痕,随年龄增长,水疱性皮损逐渐减少,而出现痒疹样损害。其余家属均无相关临床表现。
2.2 基因变异分析结果 散发患者1的COL7A1基因73号外显子存在c.6082G>A杂合,导致编码蛋白质的第2028位氨基酸由甘氨酸变成精氨酸(p.G2028R),其父母均未携带该突变(图3)。该突变已在显性营养不良型EB中报道过[2],属于致病突变。

图3 散发患者1及其父母测序结果 散发患者1携带COL7A1基因c.6082G>A突变,其父母均未携带此突变 图4 家系2先证者及其父母测序结果 先证者及其父亲同时携带COL7A1基因c.6235G>A突变和KRT5基因c.499G>A突变,母亲未携带这2个基因突变
家系2先证者父亲的全外显子组测序结果显示杂合2个基因的错义突变,即COL7A1基因c.6235G>A(p.G2079R)突变和KRT5基因 c.499G>A(p.E167K)突变,经Sanger测序先证者也携带这2个突变,其母亲无这些突变(图4),证实先证者这2个基因的突变均来自其父亲。本研究中COL7A1基因和KRT5基因的错义突变分别在显性营养不良型EB[3]和单纯型EBS[4]中报道过,均属于致病突变。
所有测序结果均通过反向测序验证,且在与医院自有的364个正常对照的全外显子组测序数据中不存在这些突变。
3 讨论
EB根据超微结构下水疱在皮肤基底膜带发生的位置不同,最新的分类将其分为单纯型EB(EBS)、交界型EB(JEB)、营养不良型EB(DEB)和kindle EB[5]。DEB的临床特征为反复发生的水疱、痒疹样结节、粟丘疹和萎缩性瘢痕,其水疱位于真皮基底膜带致密板以下,位置深在,所以愈合后留有明显的瘢痕[6]。
DEB均由编码Ⅶ型胶原的COL7A1基因突变所致,本研究中2例患者的COL7A1基因突变既往报道均导致显性遗传的痒疹样营养不良型大疱性表皮松解症(dystrophic epidermolysis bullosa pruriginosa,DEB-Pr)。李桐等[7]在2014年于国内首次报道DEB-Pr中COL7A1基因p.G2079R该突变位点,文中先证者的表型与家系2先证者父亲的表型相同。DEB-Pr于1994年首次由McGrath等[8]提出,其特征是好发于四肢伸侧的痒疹样结节、苔藓样斑块及紫罗兰色线状瘢痕,伴剧烈瘙痒和指趾甲损害[9]。DEB-Pr常于婴儿早期或儿童期发病,愈后留有白色丘疹样瘢痕,伴粟丘疹形成和甲营养不良;少数病例于成年后发病,最晚有71岁发病的报道[10]。
COL7A1基因定位于在3p21,由118个外显子组成,编码Ⅶ型胶原蛋白,由Ⅶ型胶原构成的锚原纤维是基底膜的重要组成部分[11]。Ⅶ型胶原包括中央三螺旋杆状胶原区域以及氨基端和羧基端,其中三螺旋区的甘氨酸替代的错义突变为DEBP最常见的基因突变类型,Ⅶ型胶原蛋白结构特征是Gly-X-Y的连续重复[12]。甘氨酸是最小的氨基酸且位于三螺旋区的中央轴,此位置的蛋白链紧密折叠,只能容纳最小的甘氨酸,所以甘氨酸残基的替换会扭曲三螺旋的构象,胶原合成障碍后使锚原纤维数量或结构上发生异常,进而真表皮处的连接功能减弱、皮肤脆性增加,致密板下水疱形成[13]。本研究中2例患者的COL7A1基因突变位点p.G2028R和p.G2079R为发生在不同外显子上的相同碱基替换,均为鸟嘌呤变成腺嘌呤,且均导致编码的氨基酸由甘氨酸变成精氨酸,属于经典的COL7A1三螺旋区内的甘氨酸置换,但发病时间完全不同,临床表型和发病时间不仅取决于致病突变的性质,还受细胞类型、微环境和外部因素的影响[14]。COL7A1基因型和表型的关系尚不明确,该基因同一突变位点亦有较大的临床异质性。Nakamura等[14]总结分析3例COL7A1基因中p.G2028R甘氨酸替代突变导致DEB,其表型有单纯的甲营养不良、典型的DDEB表型和DEB-Pr的表型,首次证实相同的COL7A1基因甘氨酸替换可引起明显的表型异质性。
角蛋白KRT5由基底层角质形成细胞表达,其中间丝蛋白基因突变会引起细胞骨架结构不稳定[15]。家系2中KRT5基因突变既往报道导致局限性EBS,表现为局限于手足部位反复发作的表皮内大疱,不累及黏膜和指甲,且愈后不留瘢痕[5],但家系2的先证者及其父亲从未出现过手足等部位的紧张性大疱,无既往报道的相关临床表型。角蛋白的结构从氨基端到羧基端分头区、杆区和尾区三个部分,本研究中KRT5基因的错义突变导致167位的酸性谷氨酸被碱性赖氨酸取代,突变位于角蛋白头区H1的倒数第二个氨基酸,导致角蛋白丝组装中断,削弱角质形成细胞抵抗机械应力的能力而引起水疱,角蛋白不同结构区域的突变与EBS的严重性密切相关,发生于头区H1的突变常引起轻微的临床症状,可在超微结构上呈现出相对正常的角蛋白中间丝[16]。
家系2的先证者及其父亲同时携带2个基因的错义突变:COL7A1基因的c.6235G>A和KRT5基因的c.499G>A,杂合这2个基因的错义突变尚未见报道。家系2未行透射电镜、免疫荧光等检测来定位裂隙水平,因此究竟是两个基因突变还是单个基因突变发挥作用、家系2是DEB还是DEB和EBS两亚型复合的EB类型均有待探究。 Vahidnezhad等[17]报道了两基因共遗传引起不同亚型共存的EB病例,先证者是EXPH5和COL17A1双基因纯合突变导致的伴发单纯性和交界性的EB,在组织病理学和基因水平上同时具有EBS和JEB这2种不同类型的特点。本研究中先证者表现为手足白色粟丘疹、水疱及中指指甲脱落,尚无双胫前痒疹样结节,但其父表现出DDEB的临床表型,家系2先证者随访半年未曾出现手掌和足趾等部位的大疱,并未出现EBS的临床表型,这2个基因突变的共显性遗传并没有加重每个基因单独突变的后果,推测与KRT5基因该突变位点位于角蛋白头区H1本身可导致十分轻微的临床症状且EBS的皮损会随年龄增长而改善有关。
综上所述,2例患者经基因检测的辅助诊断最终确诊为EB,家系2杂合COL7A1基因和KRT5基因的错义突变,同时携带这2个基因的杂合突变在国内外均为首报,基因检测的结果拓展了EB的突变数据库,但EB的临床表型和基因型之间的关系仍有待于进一步研究。
