丙硫菌唑关键中间体的清洁制备工艺
2022-07-14陈波
陈 波
(成都兰一科技发展有限公司,四川成都,610000)
1 前言
随着国家对环境保护要求的日益提高,优化或开发丙硫菌唑的清洁合成工艺具有非常重要的意义。我们通过不断实验改进,以a-乙酰基-r-丁内酯为原料,经过氯气氯化、盐酸开环、氢氧化钠关环、氯气再氯化得到关键中间体1-氯-1-氯乙酰基环丙烷,充分利用了体系产生的氯化氢以及工艺水的循环使用,大大减少了工艺废水的排放总量,简化了操作步骤,有效提高了合成收率,并且反应均为常规合成工艺,操作简单。
文献报道1-氯-1-氯乙酰基环丙烷的主流合成路线如下:
(1)一次氯化,a-氯-a乙酰基-r-丁内酯在低温无溶剂条件下通入氯气反应生成a-氯-a乙酰基-r-丁内酯。

(2)盐酸酸解,a-氯-a乙酰基-r-丁内酯在浓盐酸存在下加热酸解生成3,5-二氯-2-戊酮。

(3)碱水条件环合,3,5-二氯-2-戊酮在氢氧化钠水溶液中加热环合得到1-氯-1-乙酰基环丙烷。

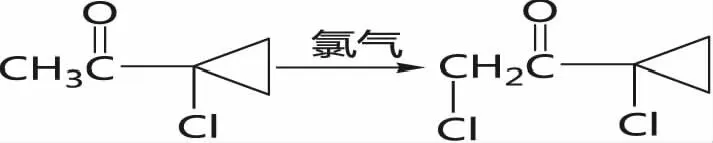
(4)二次氯化,1-氯-1-乙酰基环丙烷在低温条件下通入氯气得到1-氯-1-氯乙酰基环丙烷(丙硫菌唑关键中间体)。
2 实验部分
2.1 仪器与试剂
仪器:LC-20AT型高效液相色谱仪(岛津公司);HP6890-5973型气相-质谱联用仪(Perkin Elmer公司);Varian INOVA-300型核磁共振仪(CDC13为溶剂,TMS四甲基硅烷为内标,美国PE公司)。
原料与试剂:a-乙酰基-r-丁内酯(分析纯,浙江联化化学);C12(99%,自制);甲醇(工业级,宁夏庆华);盐酸(30%工业级,公司副产);四丁基溴化铵(工业级,常州启迪化工);氢氧化钠(99%,内蒙君正);碳酸氢钠(分析纯,山东宝臻化学);纯净水(市售)。
2.2 实验部分
2.2.1 中间体I的合成
将a-乙酰基-r-丁内酯(640g,5.0 mol)加入2 000 mL三口瓶中,冷却至0℃。慢慢通入氯气(362.5g,5.1 mol),温度控制5℃以下,尾气采用三级吸收副产工艺套用盐酸,作为本工艺中间体Ⅱ合成的原料使用,通气完成后搅拌一小时,取样经液相色谱分析原料转化完全,不需处理直接进入中间体Ⅱ的合成。
2.2.2 中间体Ⅱ的合成
在装有回流分水装置的2000 mL三口瓶中,加入回收工艺副产20%盐酸(1825 g,10mol),加热到100℃,慢慢滴加上述一批中间体Ⅰ,维持100℃滴加并在2小时滴加完成,滴加过程中无色油状中间体Ⅱ不断蒸馏出,通过分油器不断分出油层,滴加完毕开始蒸馏,约2小时蒸馏温度上升到120℃,几乎没有油状馏分蒸出,切换蒸馏馏分,后期约1000 g 5%左右稀盐酸蒸馏出的馏分作为尾气吸收的补充水,继续蒸馏约800 g 20%左右盐酸,循环套用到本工序下批投料,得到730 g无色油状物,纯度97.2%,两步连续套用综合产率91.6%。
2.2.3 中间体Ⅲ的合成
在2000mL带有回流分水装置的三口瓶中,加入回收水1000 mL、片状氢氧化钠(220 g,5.5mol)、四丁基溴化铵(3.3 g, 0.01 mol),加热到100℃,慢慢滴加中间体II(730 g, 4.58 mol),约一小时滴完,滴加过程中将生成的中间体Ⅲ通过油水分离器从反应体系分出,常压蒸馏到110℃,收集水蒸气蒸馏的油层与水层馏分进行分液,得到490 g淡黄色油状物,GC检测纯度97.0%,产率87.6%。
2.2.4 中间体Ⅳ的合成
将中间体Ⅲ(490 g, 4.01mol)、5mL甲醇加入1000 mL带有自制搅拌器的玻璃三口瓶中,冷却至0℃。慢慢通入氯气(284 g, 4.0 mol),尾气进入副产盐酸尾气吸收系统,温度控制低于5℃以下,大约5个小时完成通入氯气,保温搅拌一小时,取样分析中间体Ⅲ转化率达到99%,加入冰水1000mL,分出水层作为尾气吸收补充水,油层用饱和碳酸氢钠溶液中和至中性,分液,水层进入废水处理站,有机相减压蒸馏脱去水分,得到560.89g淡黄色油状物2-氯-1-(1-氯环丙基)乙酮,纯度93.0%,收率85.01%。
1H NMR (400 MHz, CDC13) δ 4.70 (s, 2H), 1.69 (q, J=5.0 Hz, 2H), 1.39 (m, 2H).LC-MS calcd for C5H6Cl2O (M+1)+ 152.98; found 153.10。
3 结果与讨论
(1)从a乙酰基-r-丁内酯合成中间体Ⅰ的过程中,利用已知文献资料最优温度与投料摩尔比进行此操作,在后处理时不经过水洗直接将反应混合液进入酸解过程,减少了操作程序,并避免了水洗时产品在水中的微量溶解导致物料损失。
(2)从a-氯-a乙酰基-r-丁内酯酸解合成中间体Ⅱ 3,5-二氯-2-戊酮过程,套用回收盐酸进行酸解,将因氯化氢尾气带出微量产品套回到反应体系,蒸馏产生的稀酸水套用补给尾气吸收水,既合理利用了系统产生的氯化氢与水,又将尾气系统和水层中溶解的产品回收到反应体系,较大程度地提高了最终酸解的收率,对工业化清洁生产和降低原料成本都具有较大的实际意义;另外,工艺过程采用边滴加边蒸馏的方式,中间体Ⅰ未经过后处理含有氯化氢,随着产品与水的蒸馏,维持了酸解时酸浓度的相对稳定,并且及时蒸馏出中间体Ⅱ,避免了中间体Ⅱ长时间滞留反应体系产生新的副反应杂质。
(3)从中间体Ⅱ3,5-二氯-2-戊酮在碱性条件环合过程制备中间体Ⅲ 3,5-二氯-2-戊酮,本文采用边滴加边蒸馏的方式及时将反应生产的中间体Ⅲ从反应体系经过精馏柱分馏后移除,利于反应彻底进行,通过准确地控制碱水和中间体Ⅱ的滴加速度,确保体系的碱浓度始终维持在比较稳定的略过量范围,有效地避免了因碱水浓度过高和产物在反应体系滞留时间过长而产生的副反应杂质过多;蒸馏分出的水层因溶解微量的产物中间体Ⅲ,经过循环使用,有效地减少了工艺废水的处理量,并从中回收了中间体,提高了收率,对工业生产转化时清洁生产与降低生产成本都具有积极意义。
(4)中间体Ⅲ经过再次氯化过程,继续用氯气低温氯化,并加入催化剂甲醇,在特制搅拌器和良好的搅拌体系下,可以避免多氯化副反应的发生,反应生成的氯化氢气体经过尾气吸收产生合格含量的副产盐酸,以及后处理水洗的稀盐酸套用到尾气吸收补充水产生副产盐酸,副产盐酸套用到本工艺中间体Ⅱ生产的酸解工序,有效地减少了工艺废水的排放,并充分利用了体系产生的氯化氢,而且回收了溶解在水层的中间体,因为副反应多氯化的良好控制,生产产物质量明显提高,不需要精馏含量直接达到93%以上,高于文献经过简单精馏后的92%含量,收率也因副反应减少而较文献收率提高了5%以上。
4 结论
在现有文献报道的基础上,本文经过大量的实验验证,从第一次氯化、酸解、环合到第二次氯化制备中间体1-氯-1-氯乙酰基环丙烷的过程中,通过对各工序尾气氯化氢的充分回收利用与工艺水的循环套用,不但可以降低生产成本并大量有效减少三废排放,而且实现了制备过程的清洁工艺,成功避免了文献合成中尾气处理二次污染量大、生产收率低和有机杂质进入工艺水增加废水处理难度的工艺现状,在中间体生产转化过程中为清洁生产与降低原料成本起到了重要的作用。
