GC-MS法测定保健食品中他汀类药物含量的不确定度评定
2022-07-08张丽霞郭庆园华启诚李向梅
张丽霞 郭庆园 华启诚 李向梅
(1. 华南农业大学食品学院,广东 广州 510000;2. 清远海关综合技术服务中心,广东 清远 511515;3. 广州海关技术中心,广东 广州 510000)
他汀类物质是减肥类保健食品中常检出的非法添加化合物[1]。其作用机理是通过阻断羟甲戊酸的代谢途径,增加细胞膜表面低密度脂蛋白受体的活性,致使血清中胆固醇被加速清除,从而达到降脂减肥的效果[2-3]。但过量食用他汀类化合物不仅会引发横纹肌溶解等肌肉类疾病[4]还会导致急性肾衰[5-6]。常见的非法添加他汀类物质包括洛伐他汀、美伐他汀及辛伐他汀等[7-8]。
目前,国家市场监督管理总局规定的他汀类物质检测方法为提取食品中的他汀类物质后直接用LC-MS/MS进行测定[9],该方法未涉及样品测定前的净化步骤。孙丽萍等[10]将提取后的他汀类样品直接进行HPLC测定然后评定该方法的合成不确定度,结果表明重复性分量拟合的相对标准不确定度占比54%。保健食品的基质复杂[11],适当净化可提高检测结果的精密度,同时还能规避样品基质对仪器的污染。水—氯化钠盐析法常用于去除果蔬农药残留中水分等物质[12],因此在他汀类物质检测方法的前处理过程引入水—氯化钠盐析步骤可以有效地溶解保健食品中色素、糖类等干扰物质。研究拟采用水—氯化钠盐析法对保健食品提取物进行净化,然后利用GC-MS检测美伐他汀、洛伐他汀及辛伐他汀含量,根据CNAS-GL 06—2019《化学分析中不确定度的评估指南》和JJF 1059.1—2012《测量不确定度评定与表示》的要求并结合文献[13-16],建立相关数学模型,分析及评定各不确定度分量的来源,最后利用不确定度评定对水—氯化钠前处理保健食品的检测方法进行评价分析。
1 材料与方法
1.1 仪器与试剂
美伐他汀、洛伐他汀、辛伐他汀:100 mg/L,中国First Standard公司;
乙腈、丙酮:色谱纯,美国Honeywell公司;
气相色谱质谱联用仪:GCMS-QP 2010 Ultra型,日本岛津公司;
电子天平:CPA225D型,赛多利斯科学仪器有限公司。
1.2 样品前处理方法
称取样品2.00 g,加入乙腈10.0 mL,超声15 min后过滤至含有5 mL水和2 g氯化钠的离心管中,漩涡振荡后静置5 min,分取5.00 mL乙腈上层液,80 ℃氮吹至干,1.00 mL丙酮复溶,过0.22 μm尼龙滤膜,备上机。外标法定量。
1.3 标准溶液的配制
1.3.1 混合标准工作液 分别准确量取美伐他汀、洛伐他汀及辛伐他汀储备液(100 mg/L)各0.50 mL,合并后使用丙酮定容至10.0 mL容量瓶;混合标准工作液质量浓度为5 mg/L。
1.3.2 混合标准工作系列 从5 mg/L混合标准工作液中分取10,50,100,500,1 000 μL;以丙酮分别定容至5.0 mL容量瓶;此时标准系列质量浓度为0.01,0.05,0.10,0.50,1.00 mg/L。
1.4 测定条件
1.4.1 色谱条件 进样口温度270 ℃;不分流进样;进样量1 μL;Rxi-5ms色谱柱(30 m×0.25 mm,0.25 μm),柱温:初温100 ℃,保持2 min,然后以15 ℃/min升至295 ℃,保持2 min;载气:氦气(纯度≥99.99%);恒压模式:120 kPa。
1.4.2 质谱条件 电离模式为电子轰击源(EI),能量为70 eV,离子源温度220 ℃,接口温度290 ℃;扫描方式:离子扫描(SIM)。主要色谱及质谱参数详见表1。
1.5 标准不确定度分量的评定公式
按式(1)计算合成相对标准不确定度。
(1)
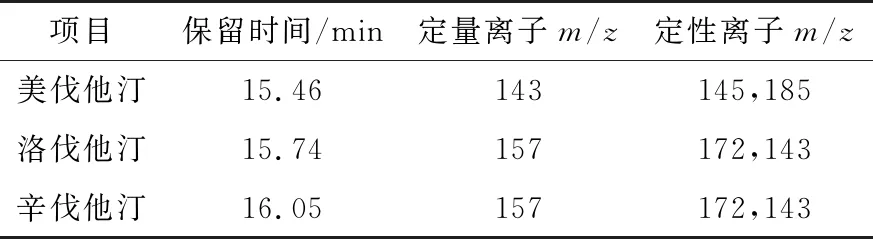
表1 保留时间及定量离子信息
式中:
uc——合成相对标准不确定度;

按式(2)计算相对标准不确定度。
(2)
式中:
urel——相对标准不确定度;
x——样品质量浓度测定值。
按式(3)计算扩展不确定度。
U=kuc(x),
(3)
式中:
U——扩展不确定度;
k——常数(95%置信水平),取2。
2 结果与分析
2.1 数学模型的建立
(4)
式中:
ω——样品中目标物的含量,mg/kg;
C——样品中目标物的质量浓度,mg/L;
V——定容体积,mL;
D——稀释因子;
m——称样量,g;
frep——重复性引入的修正因子。
2.2 不确定度的来源分析
基于测定方法及数学模型,影响试验不确定度的主要分量为标准溶液配制、标准曲线拟合、定容体积、称样量以及重复性等。影响各分量的因素详见图1。
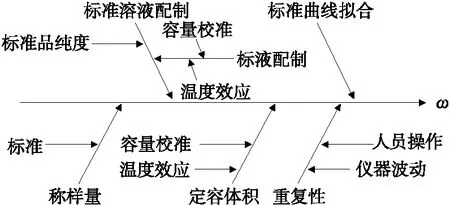
图1 不确定度来源分析图
2.3 各不确定度分量的评定
2.3.1 标准溶液配制过程引入的相对标准不确定度
(1) 标准储备液引入的相对标准不确定度:依据各项目的标准物质证书所提供的扩展不确定度U、包含因子k、标准物质浓度值X等信息,以B类评定方法计算标准储备液所引入的相对标准不确定度urel(C1),见表2。

表2 标准储备液的不确定度
(2) 配制过程引入的相对标准不确定度:依据1.3步骤,标准工作液的配制过程涉及10.0 mL容量瓶、500 μL移液器。试验使用的A级容量瓶所引入的不确定度由容量校准不确定度u(C容A)、温度效应不确定度u(C容B)两个分量组成;移液器引入的不确定度由容量校准不确定度u(C移A)、温度效应不确定度u(C移B)两个分量组成。

移液器引入的不确定度拟合方法与容量瓶一致。标准工作液使用的500 μL移液器引入的相对标准不确定度结果见表3,为2.58×10-3。

表3 定量器具引入的不确定度
1.3.2步骤中逐级稀释引入的不确定度可忽略不计。因此,按式(5)计算配制过程引入的相对标准不确定度urel(C2):
(5)
计算结果为4.18×10-3。
(3) 样品中目标物浓度引入的相对标准不确定度:按式(6)计算目标物浓度的相对标准不确定度urel(C):
(6)
计算可得美伐他汀、洛伐他汀及辛伐他汀在标准溶液配制引入的urel(C)分别为4.22×10-3,4.22×10-3,4.21×10-3。
2.3.2 标准曲线拟合所引入的相对标准不确定度 取1.3.2 配制的5个标准工作溶液分别测定3次,以峰面积A为纵坐标,标准溶液浓度Ci为横坐标拟合线性方程,结果见表4。取样品加标后上机测定,根据标准曲线回归方程计算样品中美伐他汀、洛伐他汀、辛伐他汀他的浓度,按式(7)分别计算3种他汀类物质的标准溶液浓度差的平方和:

表4 标准曲线数据†
(7)
式中:
Sxx——标准溶液浓度差的平方和;
Ci——不同标准溶液质量浓度值,mg/L;

按式(8)分别计算3种他汀类物质的标准溶液峰面积残差的标准差:
(8)
式中:
S——标准溶液峰面积残差的标准差;
Aj——标准溶液的峰面积;
B0——标准曲线的截距;
B1——标准曲线的斜率;
n——标准曲线中校准点的测量次数,n=15。
将式(7)、式(8)计算所得结果带入式(9)中计算3种他汀类物质标准曲线拟合所引入的不确定度:
(9)
式中:
u(curve)——标准曲线拟合引入的不确定度;
p——实际被测样品的重复测量次数,p=2;
x——实际被测样品的平均质量浓度值,mg/L。
计算所得美伐他汀、洛伐他汀、辛伐他汀的相对标准不确定度为1.48×10-2,1.54×10-2,2.77×10-2。
2.3.3 定容体积引入的相对标准不确定度 从10.0 mL样品的乙腈提取液中分取5.0 mL进行净化或浓缩处理,稀释因子D为2;样品提取液经80 ℃氮吹至干后,准确加入1.00 mL丙酮复溶。以上过程使用的是10,5 mL的单标线吸量管及1 000 μL移液器,其引入的不确定度拟合计算方法与2.3.1(2)容量瓶一致,计算结果详见表3。三者引入的相对标准不确定度分别为2.51×10-3,2.68×10-3,2.58×10-3。按式(5)计算定容体积引入的相对标准不确定度为4.49×10-3。
2.3.4 称样量引入的相对标准不确定度 称样量为2.00 g,依据JJG 1036—2008标准及该天平校准证书可知,其偏载误差为5.0×10-5g,k=2,则按式(10)计算称样量引入的相对标准不确定度:
(10)
式中:
d——偏载误差,g;
k——常数(95%置信水平),取2;
m——称样量,g。
计算结果为1.25×10-5。
2.3.5 重复性引入的相对标准不确定度 重复称量同一阴性样品11份,分别进行0.10 mg/kg加标,相同条件下进行前处理并测定含量值。采用A类评定,按式(11)计算重复性引入的相对标准不确定度:
(11)
式中:
urel(ωi)——重复性引入的相对标准不确定度;
S(ωi)——11份加标样品含量值的标准偏差,mg/kg;
e——加标样品的平行份数,e=11;

为考察水—氯化钠净化步骤对测定结果重复性的影响。重复称量同一阴性样品11份,分别进行0.10 mg/kg洛伐他汀加标试验。操作步骤不包括水—氯化钠盐析,其余步骤与1.2一致。11组样品的测定结果分别为0.131 1,0.109 4,0.110 3,0.120 3,0.102 2,0.104 1,0.119 0,0.120 4,0.103 2,0.130 9,0.117 2 mg/kg。此时,重复性引入的相对标准不确定度按式(11)计算,结果为2.59×10-2。其他项目重复性的测定结果见表5。

表5 各项目的重复性的测定结果
2.4 合成不确定度
汇总试验所用检测方法涉及到的相对标准不确定度分量,在此基础上,拟合95%置信区间,包含因子k=2的合成相对标准不确定度ucrel(ω)、合成不确定度uc(ω)、扩展不确定度U(ω)以及检测结果ω见表6。
由表6可知,试验所用检测方法对美伐他汀、洛伐他汀及辛伐他汀拟合的各不确定分量大小的趋势一致,主要来源是标准曲线拟合,其次是定容体积、标准溶液配制以及重复性。通过对比样品经过水—氯化钠净化与否,得出洛伐他汀引入的重复性相对标准不确定度结果:经净化后的值为3.12×10-3,未经净化后的值为2.59×10-2,前者约为后者的10%,与孙丽萍等[10]有关HPLC法测定保健食品中洛伐他汀含量的不确定度拟合分量结果一致,最后检测结果表述为(0.99±0.06) mg/g (k=2),扩展不确定度与检测值之比为6.06%。在孙丽萍[10]的研究中,不确定度的主要来源是重复性,相对标准不确定度为0.023 3,占各不确定度分量和的54%,远超过试验方法拟合的重复性分量0.003 12以及对应的占比5%。综上,若保健食品样品未经净化,样品基质干扰导致重复性引入大量的不确定度,对结果的准确性影响较大。试验所建立的前处理方法引入水—氯化钠净化步骤,有利于提高检测结果的精密度。

表6 各分量的相对标准不确定度汇总表
3 结论
对保健食品中他汀类药物进行GC-MS检测时,采用水—氯化钠净化法前处理,能够降低样品基质的干扰,减少重复性引入的不确定度,提高检测结果的精密度和准确度。重复性及样品的称取都会引入不确定度,但其主要来源于标准曲线拟合、定容体积以及标准溶液配制。因此,平时检验检测过程中要注意前处理的规范性,尽可能选用高精度以及经校准为A级量具配制溶液等。同时应定期对GC-MS进行期间核查以及检定校准,使用过程中关注仪器的状态并做好维护,以保证测定结果的可靠性。
