Vonafexor(EYP001)的合成
2022-07-05刘洪涛田文华李文燕
姜 珊, 刘洪涛, 田文华, 李文燕
(1.河北师范大学 化学与材料科学学院,河北省有机功能分子重点实验室,河北 石家庄 050024;2.河北省人民医院 药学部,河北 石家庄 050051)
Vonafexor(EYP001)是非胆酸类法尼醇X受体选择性激动剂,法尼醇X受体(famesod X receptor,FXR)是核受体超家族中的一员[1].FXR可以调控胆汁酸、脂质和糖类的代谢,影响胆淤积、糖尿病、高血脂、动脉粥样硬化和肥胖症等病理过程[2],对肾上腺、肠、肾和肝脏也有重要影响[3].有研究发现,FXR与乙型肝炎病毒(hepatitis B virus,HBV)的复制有关.HBV感染是全球性的健康问题,中国的境况更为严峻,现有临床药物难以彻底治愈慢性乙肝.共价闭合环状DNA(covalently closed circular DNA,cccDNA)是HBV感染初期能最先检测到的复制中间体,也是嗜肝DNA病毒、病毒基因和前基因组RNA的合成模板[4].在HBV的复制过程中[5],cccDNA起到了非常关键的作用,它是病毒复制的中介,在宿主肝细胞核内以微染色体的形式存在,半衰期可持续数十年,目前的治疗手段均难以将其彻底清除,导致慢性乙肝无法彻底治愈[6-7].因此,只有直接作用于共价闭合环状DNA、抑制它的形成,转录或直接封闭和降解cccDNA才能彻底清除HBV[8].目前,在进入临床的新型抗HBV的药物中,只有FXR激动剂可以有效地抑制或清除共价闭合环状DNA[9-10].非酒精性脂肪性肝炎[11-13](national association of specimeen hunters,NASH)是一种严重的非酒精性脂肪性肝病,发病率高,可发展为肝纤维化、肝硬化、肝衰竭和肝细胞癌.中国非酒精性脂肪性肝病患病率为20.1 %,已成为慢性肝病以及肝移植的主要病因.FXR激动剂可以减缓非酒精性脂肪性肝病的肝细胞脂肪变性和脂质沉积,对肝脏起到保护作用[14].Vonafexor(EYP001)是法国Eyno公司研发的一种非胆酸类选择性FXR激动剂,目前正用于慢性乙型肝炎和NASH的II期临床研究中[15].为进一步研究该化合物,本文中,笔者以5-溴水杨醛为起始原料,通过2步反应生成5-溴苯并呋喃-2-甲酸乙酯中间体1,然后1与1-Boc哌嗪反应生成中间体2,2通过氯原子取代反应得到中间体3,将3进行脱Boc反应后得到中间体4,再与2,6-二氯苯磺酰氯反应,得到中间体5,最后通过水解反应得到最终产物EYP001.目标产物总收率为58.3 %(按5-溴水杨醛计).合成路线见图1.其中,a为溴乙酸乙酯,K2CO3,NMP,65 ℃;b为DBU,EtOH,回流;c为Pd(OAc)2,BINAP,Cs2CO3,甲苯,100 ℃;d为浓H2SO4,THF,40 ℃;e为TAF,CH2Cl2;f为吡啶,THF,室温;g为THF,10 %NaOH,EtOH.
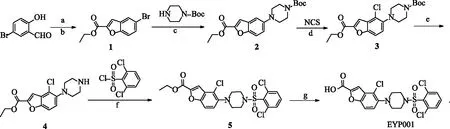
图1 目标化合物的合成路线Fig.1 Synthetic Route of Target Compound
1 实验部分
1.1 仪器及试剂
薄层监测用2F-20D暗箱式紫外分析仪(巩义市予华仪器有限责任公司);质谱用3200 QTRAP 1200 infinity series质谱仪(美国 AB Sciesx 公司);高分辨质谱用AB Sciesx TripleTOF 5600+(美国 AB Sciesx 公司);核磁共振用WIPM-NMR-400核磁共振波谱仪(中科牛津波谱技术有限公司),四甲基硅烷(TMS)为内标.
化学试剂均为市售、分析纯.
1.2 实验步骤
1.2.1 5-溴苯并呋喃-2-甲酸乙酯(1)的制备
取干燥的50 mL茄形瓶,加入5-溴-2-羟基苯甲醛2.00 g(10.00 mmol),加入20 mL的N-甲基吡咯烷酮溶解,再向反应瓶中加入K2CO32.76 g(20.00 mmol),室温下搅拌 20 min后向反应瓶中滴加溴乙酸乙酯1.60 mL(14.00 mmol),升温至65 ℃,TLC监测反应进程,30 min后反应完全.向反应液中加入20 mL水,用乙酸乙酯萃取3次,合并有机相,用饱和食盐水洗涤,无水硫酸镁干燥.滤除干燥剂,滤液减压浓缩,得到黄色油状中间体的粗产品2.50 g;用20 mL无水乙醇溶解,在室温搅拌下缓慢向反应瓶中滴加1,8-二氮杂二环十一碳-7-烯(DBU) 2.0 mL(13.38 mmol),滴加完毕后,100 ℃加热回流,TLC监测反应进程,约1 h 后反应完全.将反应液冷却至室温,除去大部分溶剂,冰水浴下向25 mL冰水中滴加浓缩的反应液,搅拌,有固体析出,减压抽滤,用蒸馏水洗涤滤饼.真空干燥后得到中间体1(淡黄色固体)2.41 g,收率为 90 %.1HNMR(400 MHz,CDCl3)δ:7.80(d,J=1.3 Hz,1H),7.55~7.49(m,1H),7.46(s,1H),7.44(s,1H),4.44(q,J=7.1 Hz,2H),1.42(t,J=7.1 Hz,3H).13CNMR(101 MHz,CDCl3)δ:159.18,154.34,146.83,130.59,128.86,125.33,116.89,113.90,112.85,61.80,14.34.ESI-MS,m/z:269.0[M+H]+,291.2[M+Na]+.
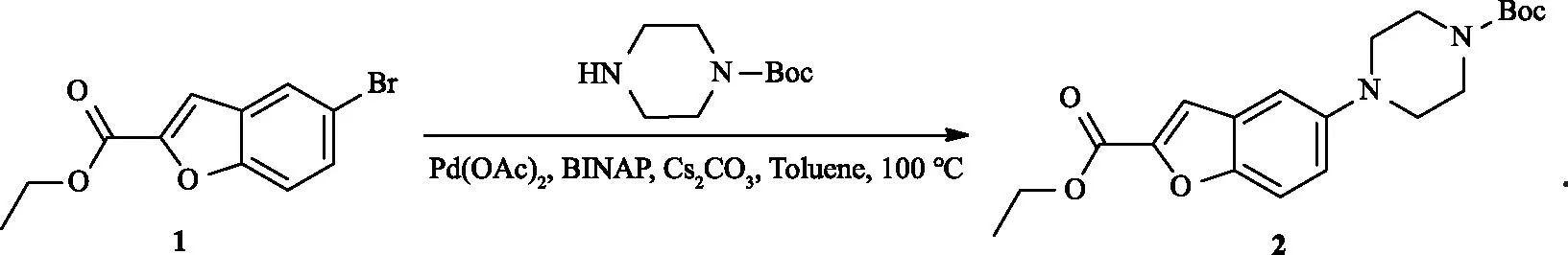
1.2.2 5-(4-叔丁氧羰基哌嗪)苯并呋喃-2-甲酸乙酯(2)的制备
将30 mL甲苯置于三颈瓶中,在室温条件下,用流动的氮气做保护,加入40 mg醋酸钯,搅拌溶解后加入0.11 g BINAP,6.26 g(19.27 mmol)碳酸铯,搅拌20 min,再加入2.40 g(9.64 mmol)中间体1和3.13 g(10.60 mmol)1-Boc哌嗪,搅拌10 min 后升温至100 ℃.TLC监测反应进程,7 h后反应结束.将反应液冷却至室温,向反应液中加入50 mL水,用乙酸乙酯萃取3次,合并有机相,依次用水、饱和食盐水洗涤,无水硫酸钠干燥;次日减压抽滤,滤液减压浓缩后进行柱层析分离(V(石油醚)∶V(乙酸乙酯)=10∶1),得到中间体2(淡黄色固体)2.85 g,收率为85 %.1HNMR(400 MHz,CDCl3)δ:7.45(d,J=9.1Hz,1H),7.41(s,1H),7.13(d,J=9.1Hz,1H),7.07(s,1H),4.40(q,J=7.1 Hz,2H),3.62~3.54(m,4H),3.07(s,4H),1.46(s,9H),1.39(t,J= 7.1 Hz,3H).13CNMR(101 MHz,CDCl3)δ:159.51,154.68,151.27,148.73,146.24,127.57,120.41,113.73,112.67,109.04,79.86,61.36,51.07,28.44,14.31.ESI-MS,m/z:375.1[M+H]+,397.2[M+Na]+.
1.2.3 4-氯-5-(4-叔丁氧羰基哌嗪)苯并呋喃-2-甲酸乙酯(3)的制备
将2.80 g(10.78 mmol)中间体2加入到30 mL四氢呋喃中,搅拌溶解后加入2.80 mL浓硫酸,将1.54 g(11.48 mmol)N-氯代丁二酰亚胺(NCS)溶于四氢呋喃中,25 ℃下缓慢滴入反应液中,滴加完毕后,40 ℃加热反应,TLC监测反应进程,1 h后反应完全.冷却至室温后,蒸除溶剂,加水,用乙酸乙酯萃取3次,依次用水、饱和食盐水洗涤.有机相用无水硫酸镁干燥,2 h后抽滤,滤液减压浓缩后得到中间体3(白色固体)3.03 g,收率为89 %.1HNMR(400 MHz,CDCl3)δ:7.59(s,1H),7.46(d,J=8.8 Hz,1H),7.21(d,J=8.6 Hz,1H),4.44(q,J=7.1 Hz,2H),3.67~3.60(m,4H),3.01(s,4H),1.49(s,9H),1.43(t,J=7.2 Hz,3H).13CNMR(101 MHz,CDCl3)δ:159.06,154.80,152.01,146.60,145.21,128.07,121.62,120.59,112.42,110.97,79.77,61.63,51.78,28.43,14.26.
ESI-MS,m/z:408.7[M+H]+,430.9[M+Na]+.
1.2.4 4-氯-5-(哌嗪基)苯并呋喃-2-甲酸乙酯(4)的制备
将3.00 g中间体3溶于15 mL二氯甲烷中,向反应瓶中滴加三氟乙酸(15 mL),室温搅拌,TLC监测反应进程,20 min后反应完全.冰水浴搅拌下,用饱和碳酸钠水溶液将反应液pH调成中性.随后用二氯甲烷萃取反应液,有机相用无水硫酸镁干燥后,减压抽滤,滤液减压浓缩得到粗产品中间体4(白色固体)2.10 g,收率为93 %.此步反应未经纯化直接投入下步反应.
1.2.5 4-氯-5-[4-(2,6-二氯苯磺酰基)-1-哌嗪基]苯并呋喃-2-甲酸乙酯(5)的制备
将2.00 g中间体4粗产品加入50 mL茄形瓶中,用重蒸的四氢呋喃(25 mL)溶解,将5.76 mL(71.28 mmol)吡啶分3次加入反应瓶,每间隔1 h加入1次,每次加完吡啶后,加入1.75 g(7.12 mmol)2,6-二氯苯磺酰氯,也分3次加入.室温搅拌反应,TLC监测反应进程,7 h后反应完全.将反应液减压蒸馏除去溶剂,向反应液中加入20 mL水,用乙酸乙酯萃取3次,合并有机相依次用水、饱和食盐水洗涤.有机相用无水硫酸镁干燥后抽滤,滤液减压浓缩后进行重结晶(V(石油醚)∶V(乙酸乙酯)=4∶1),得到中间体5(白色固体)3.09 g,收率为92 %.熔点212~214 ℃.1HNMR(400 MHz,CDCl3)δ:7.57(s,1H),7.48(dd,J=13.2,8.2 Hz,3H),7.38~7.31(m,1H),7.21(d,J=8.9 Hz,1H),4.44(q,J=7.1 Hz,2H),3.68~3.60(m,4H),3.17~3.10(m,4H),1.42(t,J=7.1 Hz,3H).13CNMR(101 MHz,CDCl3)δ:159.10,152.17,146.68,144.45,135.74,134.72,131.92,131.63,128.07,121.68,111.35,111.06,61.82,51.78,51.70,51.63,14.38,14.25.ESI-MS,m/z:518.8[M+H]+,540.8[M+Na]+.
1.2.6 Vonafexor(EYP001)的合成
向50 mL 茄形瓶中加入3.00 g中间体5,四氢呋喃(50 mL)为溶剂,加入50 mL 10 % NaOH(质量分数)水溶液,室温搅拌反应,TLC监测反应进程,2 h后反应完全.减压除去反应液中大部分溶剂,冰水浴下,1 mol/L HCl溶液将反应液调至酸性,有大量的白色固体析出,减压抽滤,蒸馏水洗涤滤饼,滤饼经重结晶(V(石油醚)∶V(乙酸乙酯)=4∶1)纯化,真空干燥后得到纯品2.52 g,收率为89 %,熔点288~290 ℃.1HNMR(400 MHz,DMSO)δ:7.81~7.74(m,2H),7.68(dd,J=8.8,7.2 Hz,1H),7.61(d,J=8.8 Hz,1H),7.30(d,J=8.9 Hz,1H),7.18(s,1H),3.53(s,4H),3.15~3.04(m,4H).13CNMR(101 MHz,DMSO)δ:161.45,155.04,144.22,135.06,134.62,133.95,132.72,128.71,119.98,119.47,111.37,107.08,51.75,46.30.高分辨质谱HR-MS:C19H15Cl3N2O5SH for[M+H]+,理论计算值为488.976 7,测试数值为488.955 8.
2 反应条件的优化
碳氮偶联反应见式(1).在实验过程中,碳氮偶联反应比较难发生,反应条件苛刻.为了促进反应发生,加入催化剂膦配体.膦配体、碱、溶剂等反应条件的优化结果见表1.
(1)

表1 苯并呋喃环碳氮偶联反应条件Tab.1 C-N Coupling Reaction Conditions of Benzofuran Ring
苯并呋喃环碳氮偶联反应条件比较苛刻,该反应以醋酸钯作催化剂,加入有机膦配体,在碱性条件和氮气保护下进行反应.为了提高反应收率,对膦配体、碱、温度和溶剂等条件进行了优化.尝试了Ph3P/NaH,Ph3P/t-BuONa等组合,发现反应不能发生.换用(t-Bu)3P/t-BuONa并用甲苯作溶剂,升高反应温度后能够发生反应,但产物杂乱,没有目标产物生成.随后降低反应体系碱性,用(t-Bu)3P/Cs2CO3催化,能够得到目标产物但是转化率较低.将配体换为BINAP后,产率有所提高,分别用t-BuONa,K2CO3,Cs2CO3催化,发现Cs2CO3对反应最为有利,产率可达85 %.因此,此步反应条件最终确定为氮气保护,Pd(OAc)2/BINAP/Cs2CO3催化,100 ℃,在甲苯中反应.
3 结 论
以5-溴水杨醛为起始原料,通过环合、取代、脱保护、磺酰化、水解5步合成了EYP001,总收率为58.3 %(按5-溴水杨醛计算),并对关键反应步骤进行了优化.此路线反应时间短、操作简单且每步都有较高的收率,为化合物EYP001的进一步开发及其衍生物的合成奠定了基础.
