马克斯克鲁维酵母FIM1基因组分析
2022-07-05罗通宇曾俊源吕奕琳周峻岗
罗通宇,曾俊源,吕奕琳,周峻岗,余 垚,吕 红
(1.复旦大学 遗传工程国家重点实验室,上海 200438; 2.复旦大学 生命科学学院 上海工业菌株工程技术研究中心,上海 200438)
马克斯克鲁维酵母(Kluyveromycesmarxianus)在分类上位于真菌界-子囊菌门-酵母纲-酵母目-酵母科-克鲁维属。属名Kluyveromyces是为纪念荷兰微生物学家Albert Jan Kluyver(1888—1956);种名marxianus是为纪念其发现者Marx,他于1888年首次从葡萄中分离得到该菌种[1]。之后,人们发现K.marxianus在发酵乳制品如奶酪和开菲尔粒(Kefir grains)中大量存在,也因此为人们所熟知[2]。除了安全可食用外,K.marxianus还具备耐高温、生长快速、代谢底物广等多种优良特性,有广泛的工业应用潜力。但是目前,由于人们对K.marxianus的遗传学和生理学的认识有限,阻碍了其进一步应用[3]。因此,研究人员需要从基因组入手,更细致地研究其生理表型与基因突变、基因表达的内在联系,增加对K.marxianus的认识。
现有对K.marxianus基因组的研究主要集中在基因组测序组装与注释方面,目前仍然缺乏对K.marxianus种内分化、核心基因鉴定的研究。截至2021年3月,美国国家生物信息中心(NCBI)上已有17株K.marxianus的基因组信息,其中5株组装出完整的染色体,在此基础上,有3株K.marxianus完成了基因组注释工作。2015年,Lertwattanasakul等人使用鸟枪法完成了K.marxianusDMKU3-1042(简称DMKU3-1042)的基因组测序、组装与注释工作[4]。同年4月,Inokuma等人完成了对K.marxianusNBRC1777(简称NBRC1777)的基因组测序、组装与注释工作[5]。2019年10月,本实验室完成了K.marxianusFIM1(简称为FIM1)的基因组测序、组装与注释工作[6],并将结果提交到了NCBI,编号为GCA_001854445.2。但后续应用FIM1的基因组信息进行深入地遗传学研究时发现,对FIM1部分位点进行Sanger法测序,重测序结果与提交到NCBI上的对应序列不符。这种问题的存在不利于后续对其进行遗传学研究与遗传工程改造,所以需要对FIM1的基因组序列及注释进行校验更新。
综上,研究从FIM1出发,使用重测序的DNA-seq数据校验了FIM1的基因组序列,随后补充了FIM1的注释,并使用比较基因组学的方法进一步完善了FIM1的基因组信息。将DNA-seq比对到之前已完成组装的FIM1基因组上,根据比对结果校验了FIM1的基因组序列。随后,研究评估了FIM1基因组质量,补充了FIM1的非编码RNA与次级代谢产物基因簇注释信息。进一步,研究对FIM1进行了比较基因组学研究。对FIM1与DMKU3-1042、乳酸克鲁维酵母KluyveromyceslactisNRRL Y-1140、酿酒酵母SaccharomycescerevisiaeS288C共3株酵母进行基因组共线性分析,划定了FIM1在酵母分化过程中的基因排列顺序保守的区域。对FIM1与NCBI来源的11株K.marxianus进行种内基因组比较,分析了FIM1与其他完成组装的K.marxianus的全基因组序列差异,构建了12株酵母的同源基因家族,并依此绘制了种内系统进化树,构建了K.marxianus的泛基因组,鉴定了FIM1菌株特性基因及其涉及的代谢通路,为FIM1遗传学的研究与遗传工程的改造提供了数据支持。
1 材料与方法
1.1 材料
本文中所有基因组与注释数据均下载自NCBI。在NCBI中的“Genome”条目下输入“Kluyveromycesmarxianus”查找K.marxianus所有菌株序列信息,下载12株基因组组装到Contig水平的K.marxianus基因组与注释信息。乳酸克鲁维酵母(K.lactis)、毕赤酵母(Pichiapastoris)、酿酒酵母(S.cerevisiae)的基因组与蛋白信息均源自NCBI上对应物种参考基因组。详细信息如表1所示。
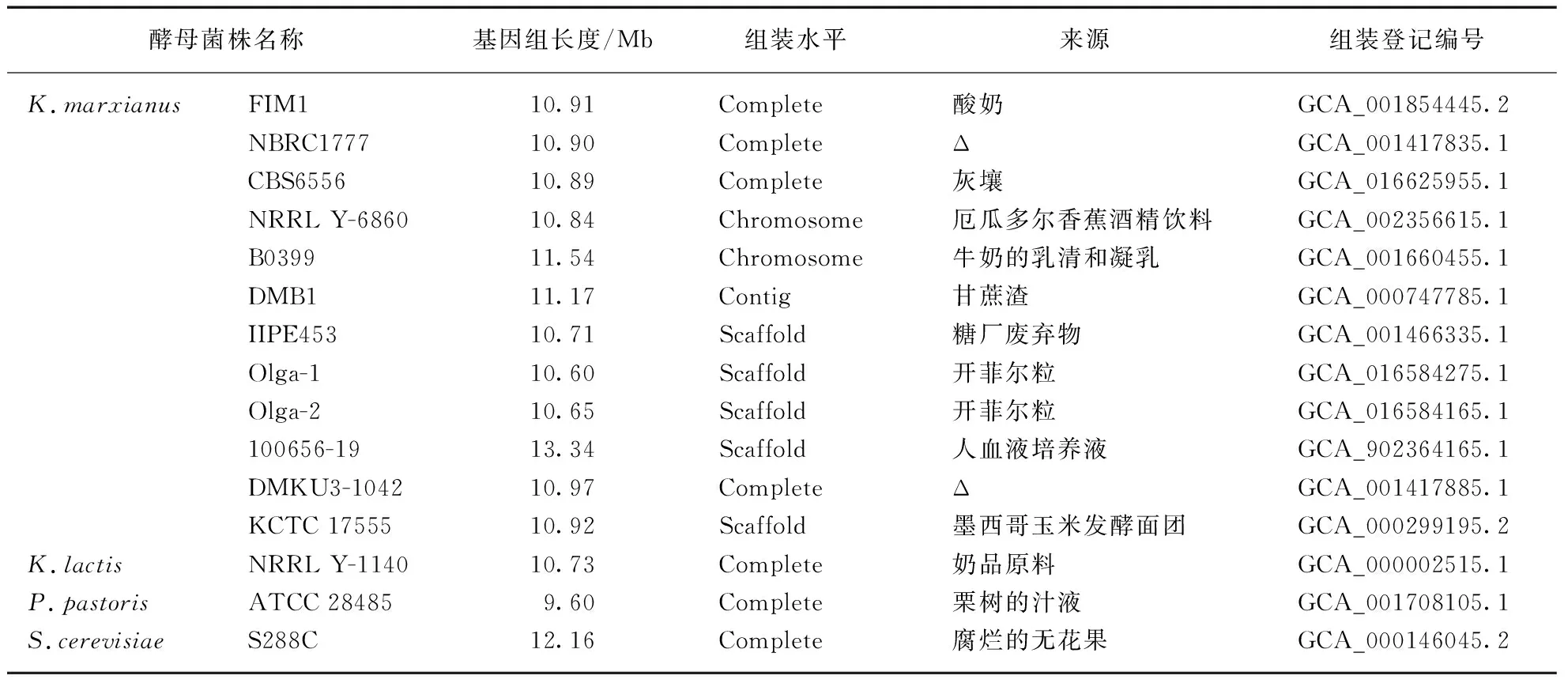
表1 本研究使用菌株的信息Tab.1 Information on strains used in this study
1.2 方法
1.2.1 FIM1基因组序列校验
研究使用抽提好的DNA基于Illumina Truseq Nano试剂构建文库,随后使用Illumina Hiseq X测序平台进行重新测序,测序深度为200×。测序结果经FastQC(https:∥www.bioinformatics.babraham.ac.uk)质量检验后,使用HISAT2[7](version 2.1)将DNA-seq数据比对到基因组上,将比对结果与FIM1基因组序列输入到inGAP[8](version 1)中,根据比对结果,删除无reads比对的区域,生成新的FIM1序列。
1.2.2 FIM1基因组组装质量评估
使用BUSCO[9](version 5.0)中的酵母目保守基因库(saccharomycetales_odb10)与真核生物保守基因库(eukaryota_odb10)分别评估FIM1的基因组质量,运行基因组评估默认参数。用于评估的酵母目保守基因库包含2137个基因,真核生物保守基因库包含255个基因。
1.2.3 FIM1的补充注释
将Rfam[10](verison 14)及其依赖软件下载到本地后,使用Rfam的默认参数预测FIM1的非编码RNA序列位置与功能。
将antiSMASH[11](version 5.1.1)及其依赖软件下载到本地后,运行antiSMASH,输入FIM1基因组序列,分类学的运行参数选择“fungi”,其他参数为全功能运行模式默认参数。
1.2.4 基因组共线性分析
使用Python版的MCscan[12](jcvi工具包)进行共线性分析,比较FIM1与DMKU3-1042、K.lactisNRRL Y-1140、S.cerevisiaeS288C 3株酵母的基因组共线性情况。使用jcvi.formats.gff模块,输入基因组序列与注释文件获得基因组bed格式的注释信息。之后输入蛋白序列与bed文件进行共线性分析,将分析结果整理后,使用jcvi.graphics.karyotype工具包生成共线性分析结果。
1.2.5K.marxianus种内基因组序列比较
使用Mummer[13](version 4)将FIM1与其他完成组装的K.marxianus基因组序列进行比较。运行Mummer的dnadiff参数,输入FIM1与待比较的K.marxianus基因组序列,在report文件中获取基因组差异比较结果。
1.2.6 系统进化树绘制及泛基因组分析
使用Orthofinder[14]进行同源基因家族分析,输入菌株的蛋白序列到Orthofinder中,然后使用DIAMOND[15]方法比对蛋白序列并构建同源基因家族,极大似然法构建系统发育树,建树的推理方法使用默认的fasttree软件,其他运行参数均为Orthofinder的默认参数。由于12株K.marxianus物种的分化时间尚不清楚,所以研究中增加K.lactis、P.pastoris、S.cerevisiae3株已经确定分化时间的酵母对进化树进行标定校正,3个物种参考分化时间通过Timetree[16]网站获取。然后使用CAFE[17](version 4.2.1)分析12株酵母在各个时间节点的基因收缩与扩增情况。随后使用r8s软件[18]绘制K.marxianus种内系统进化树,在iTOL[19]中将用于时间标定的3株酵母的信息删除。最后使用PanGP[20]预测并构建K.marxianus的泛基因组。
除FIM1蛋白序列外,本研究用到的蛋白序列由以下3种方法获得。(1) 从NCBI上下载NBRC1777、DMKU3-1042、K.lactisNRRL Y-1140、S.cerevisiaeS288C、ATCC 28485共5株酵母的蛋白序列。(2) 输入基因组序列与注释信息到Cufflinks[21]中,使用gffreads模块提取Olga1、Olga2的蛋白序列。(3) 使用AUGUSTUS[22](version 3.3)软件预测基因,运行getAnnoFasta.pl脚本提取CBS6556、NRRL Y-6860、B0399、DMB1、IIPE453、100656-19、KCTC 17555共7株K.marxianus的蛋白序列。为了优化预测结果,运行AUGUSTUS前,使用DMKU3-1042的基因组信息对AUGUSTUS进行训练。
1.2.7 FIM1特有基因的GO富集分析
通过Orthofinder中生成的Orthogroups_UnassignedGenes.csv结果文件,获得FIM1的特有基因信息。随后使用R语言中的clusterprofiler[23]程辑包配合org.Sc.sgd.db物种数据库(http:∥www.bioconductor.org/packages/release/data/annotation/html/org.Sc.sgd.db.html),输入FIM1中有功能注释的特有基因,进行GO富集分析。
2 结 果
2.1 FIM1基因组序列校验及质量评估
本研究使用HISAT2将重测序的DNA-seq数据比对到FIM1基因组上,inGAP可视化比对结果(图1,见第306页),根据比对结果删除DNA-seq无法覆盖的序列片段61处,其中基因外部片段44处,基因内部片段17处,无基因启动子或终止子被删除,删除序列的详细信息见表2(见第306页)。FIM1与校验前相比,共删除序列4 910 bp,新序列长度为10 909 543 bp。为了评估校验是否提高了FIM1基因序列的精度,使用BUSCO评估校验前后的FIM1基因组质量。采用BUSCO构建的真核生物保守基因库(eukaryota_odb10)与酵母目保守基因库(saccharomycetales_odb10)对FIM1基因组质量进行了评估(表3,见第307页),结果显示校验后基因组质量未发生改变。评估显示,FIM1基因组序列可覆盖真核生物保守基因库中91.4%的基因,与酵母目保守基因库中99.5%的基因。2019年,McCarthy使用BUSCO的酵母目保守基因库(saccharomycetales_odb9)评估了NCBI上100株S.cerevisiae酵母的基因组组装质量,其中得分最高的酵母的基因组序列可覆盖酵母目保守基因库中98.8%的基因[24]。FIM1基因组组装质量比该值高0.7%,表明FIM1基因组完整度较好,可以用于后续实验与分析。

图1 FIM1基因组序列校验Fig.1 Genome sequence verification of FIM1(a) 图中红色和蓝色线条分别代表正义链和负义链双端测序的DNA-seq匹配到FIM1的CP015054.1号染色体上的结果,下方坐标指标明了染色体位置,灰色表示对应位点的DNA-seq比对结果不佳。(b) 将灰色部分的DNA-seq比对结果进行可视化,展示了具体的reads与对应位点的比对关系。
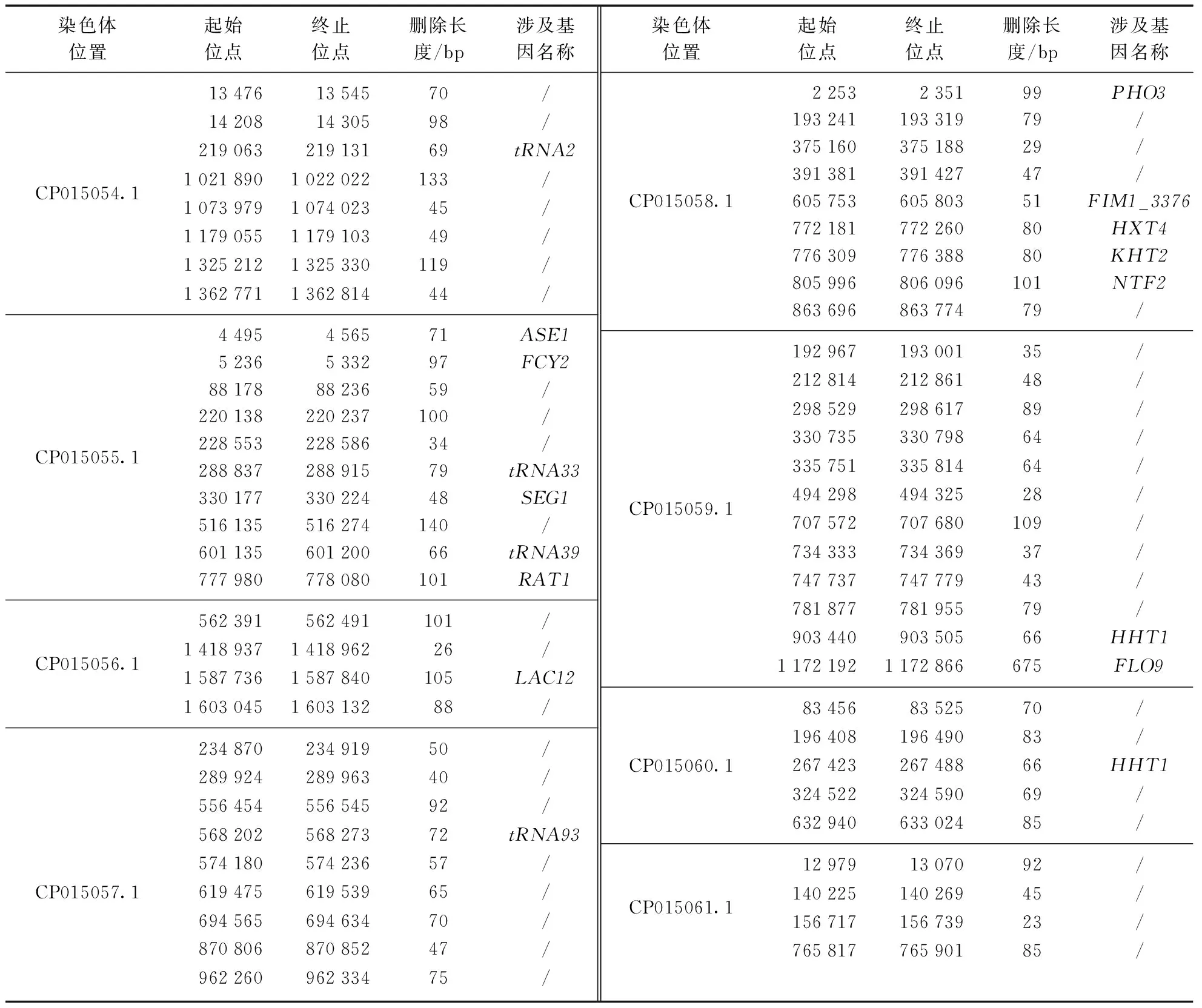
表2 FIM1删除序列的位置信息Tab.2 Position of FIM1 deletion sequence
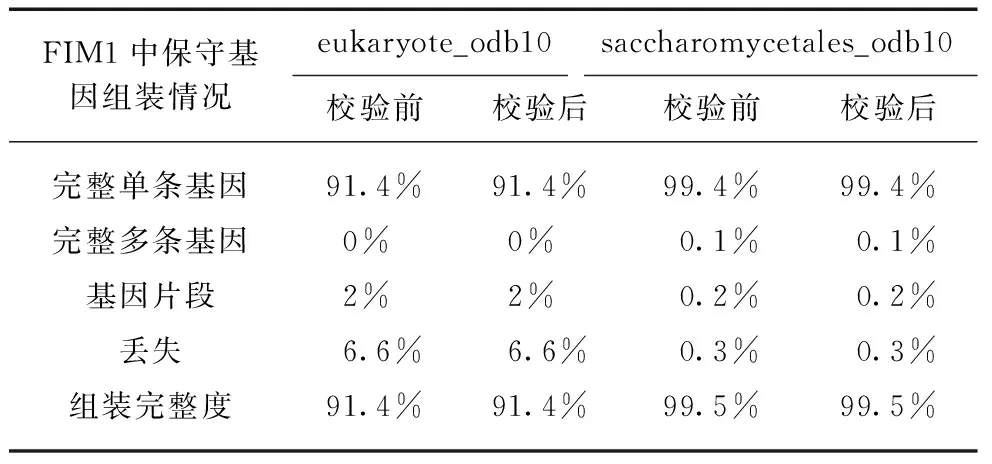
表3 FIM1基因组质量评估Tab.3 Quality assession of the FIM1 genome
为了进一步评估校验结果,研究以基因内部有序列的删除的HHT1(Histone H3)基因为例,对校验结果进行再评估。HHT1基因编码的蛋白为H3组蛋白,是一种高度保守的蛋白,在FIM1与DMKU3-1042中均有两个拷贝,校验前FIM1中H3组蛋白的长度为158 aa(amino acids),而校验后长度为136 aa。将校验前后FIM1的H3组蛋白序列与DMKU3-1042的H3组蛋白序列进行比对,结果显示校验后FIM1的H3序列与DMKU3-1042的H3序列匹配率为100%,而校验前FIM1中的H3组蛋白序列与DMKU3-1042相比有22 aa重复(图2),说明DNA-seq数据校验提高了FIM1基因组序列的质量。

图2 FIM1与DMKU3-1042的H3蛋白序列比对Fig.2 Sequence alignment of H3 between FIM1 and DMKU3-1042KLMA_60129、KLMA_80119为DMKU3-1042的H3蛋白序列,FIM1_4163、FIM1_4900为FIM1的H3蛋白序列。
2.2 FIM1基因组补充注释
使用Rfam注释FIM1基因组的非编码RNA,结果显示FIM1基因组中含有tRNA 154个,snRNA(Small nucleolar RNA)55个,rRNA 8个。将Rfam预测的结果与FIM1原有注释合并,整合后预测FIM1含有tRNA 172个,其他非编码RNA数目与Rfam预测结果一致。
使用antiSMASH注释FIM1次级代谢产物基因簇信息,结果显示FIM1有一个次级代谢产物基因簇,为非核糖体肽合成酶簇片段(NRPS-like),基因簇总长44 161 bp(表4)。

表4 非核糖体肽合成酶簇片段信息Tab.4 Information of non-ribosomal peptide synthetase cluster fragment
2.3 FIM1与3株酵母的基因组共线性分析
为了探究FIM1在物种分化过程中基因组的变化,本文分别选取了与FIM1同种的DMKU3-1042、同属的K.lactisNRRL Y-1140、同科的S.cerevisiaeS288C与FIM1进行基因组共线性分析。结果显示FIM1与DMKU3-1042在基因组共线性程度极高(图3(a)),共97.8%的FIM1序列与DMKU3-1042有同源性。FIM1与DMKU3-1042相比,1号与6号染色体的序列反向,序列无大片段缺失、重复等现象。FIM1与K.lactisNRRL Y-1140(KL)进行基因组共线性分析,结果显示两者基因组共线性程度较高(图3(b)),共92.00%的FIM1序列与KL有同源性,两者分化引起了大量的片段重排,未发现大规模基因复制现象,结果提示了基因组重排可能是克鲁维属酵母的一种重要进化方式。FIM1与S.cerevisiaeS288C(SC)的共线性分析结果显示两者基因组共线性程度不高(图3(b)),共18.91%的FIM1序列与SC有同源性,FIM1的1号和8号染色体上有两个基因片段,在SC上发生了基因片段复制现象,这一结果是对酿酒酵母曾发生过全基因组复制现象的支持。共线性分析的结果划定了FIM1基因组上基因排序保守的区域,结合其他3株已有的基因功能注释,可以对FIM1的基因功能注释进行再校验。
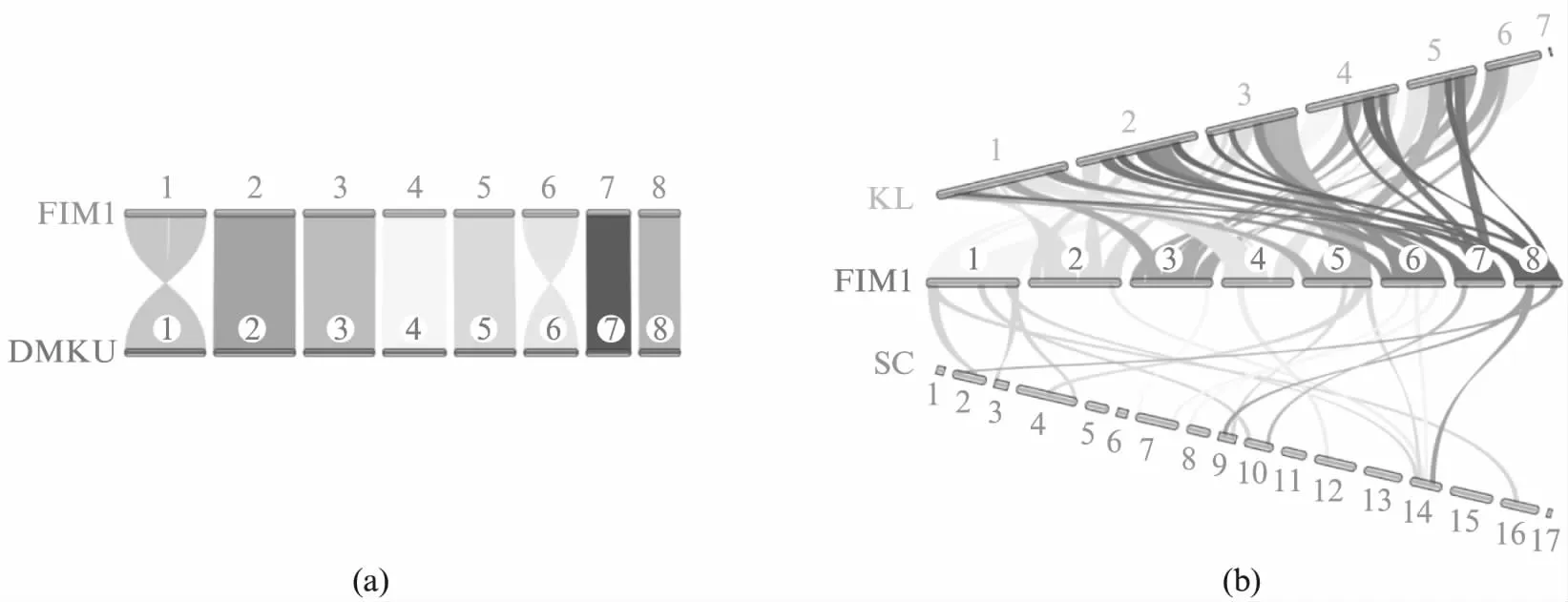
图3 FIM1基因组共线性分析Fig.3 Genome collinearity analysis of FIM1(a) FIM1与K.marxianus DMKU3-1042(DMKU)的共线性分析;(b) FIM1分别与K.lactis NRRL Y-1140(KL)、S.cerevisiae S288C(SC)的共线性分析。图中数字代表酵母的染色体编号。
2.4 K.marxianus基因组序列比较
除了FIM1外,NCBI上有DMKU3-1042、NBRC1777、CBS6556、NRRL Y-6860共4株K.marxianus组装出完整染色体信息。本文使用Mummer将这4株酵母与FIM进行基因组间序列比对分析(表5)。
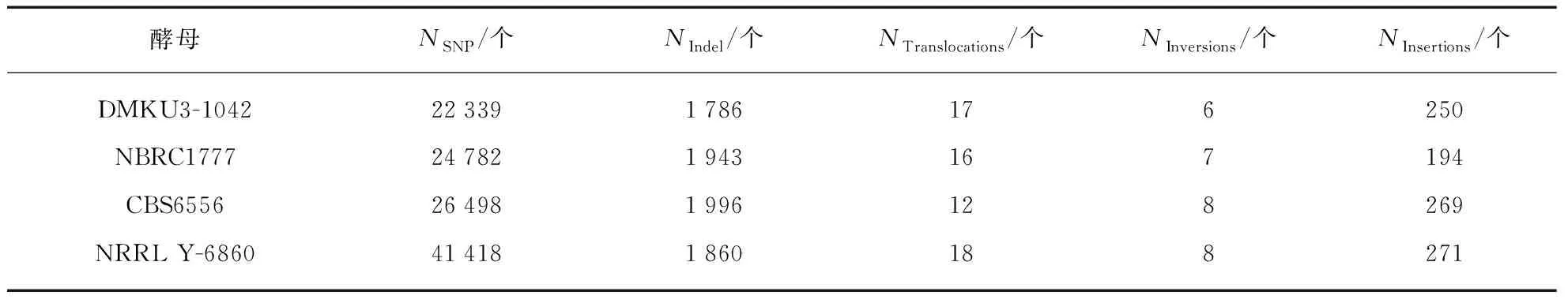
表5 FIM1与其他4株K.marxianus的基因组差异Tab.5 Genomic variation of 4 strains of K.marxianus compared with FIM1
分析结果显示,与FIM1相比,4株酵母基因组差异情况总体比较接近,基因组上产生的单碱基插入/缺失(Indels)的数目在1 786~1 996之间;易位(Translocations)的数目在12~18之间;倒位(Inversions)的数目在6~8之间;大片段插入(Insertions)的数目在194~271之间。其中只有单碱基变异(SNP)数目出现了明显差异,与FIM1相比,NRRL Y-6860出现了41 418个SNP变异,而其余3株酵母的SNP数目在22 339~26 498之间。
2.5 构建K.marxianus泛基因组
使用Orthofinder分析并构建K.marxianus的同源基因家族,分析得到5 110个同源基因家族,其中4 009个为12株酵母共有的同源基因家族。生成的结果中SpeciesTree_rooted.txt文件是软件从所有同源基因家族中推断出的STAG(Species Tree from All Genes)物种树[25],并以STRIDE(Species Tree Root Inference from Duplication Events)为根[26]。分析结果显示,12株K.marxianus早在约180万年前分化为两个亚型,第一个亚型的包含4株酵母在分化时主要发生基因扩增现象,共扩增112个基因,收缩48个基因;第二个亚型的包含8株酵母在分化时主要发生基因收缩现象,共扩增30个基因,收缩222个基因(图4)。与未分化时相比,FIM1共扩增141个基因,收缩536个基因。
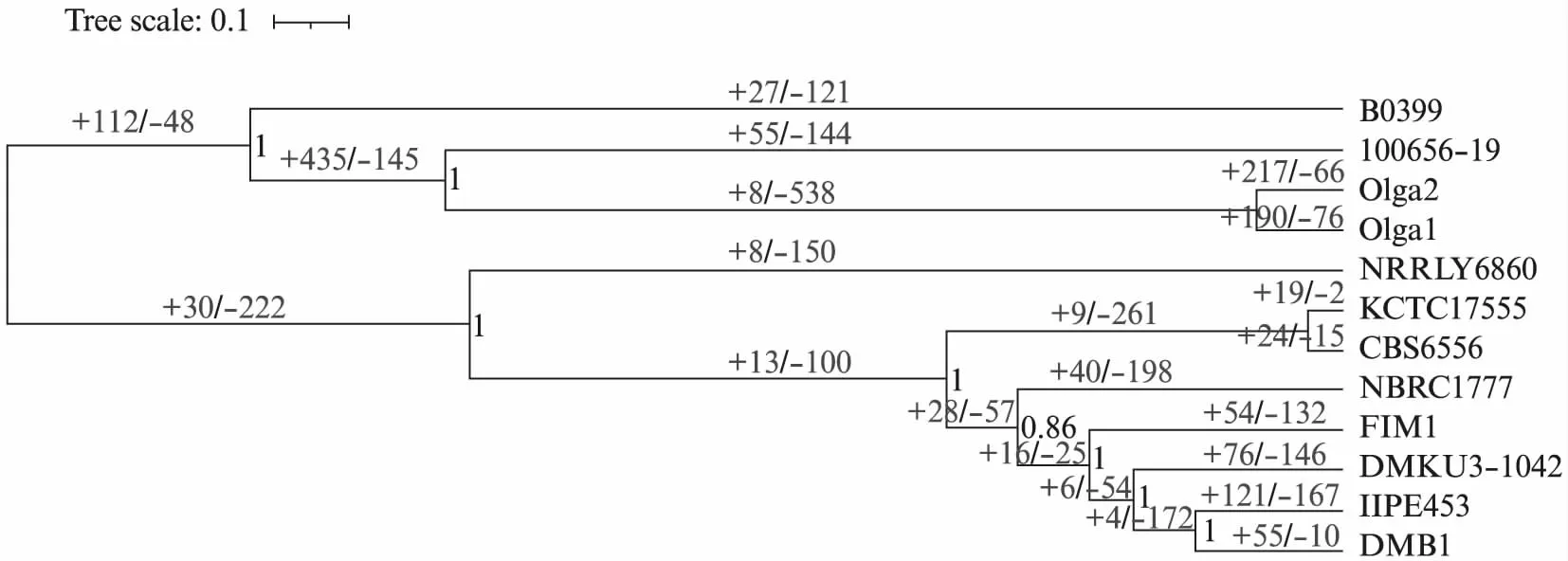
图4 K.marxianus系统进化树Fig.4 Phylogenetic tree of K.marxianus系统进化树共包含12株K.marxianus的发育信息,分化时间由3株(K.lactis、P.pastoris、S.cerevisiae)已知分化时间的酵母标定校正。获得系统发育信息后,使用iTOL将用于时间校正的3株酵母的发育信息删除。系统进化树树干上,红色代表基因扩增,蓝色代表基因收缩,进化树各个节点的support value由fasttree的SH test算法计算。Tree scale中0.1指10万年。
根据Orthofinder生成的结果(Orthogroups_UnassignedGenes.csv),获得FIM1的特有基因信息,结果显示与其他K.marxianus相比,FIM1有129个特有基因,其中有功能注释的基因有41个。将有功能注释的41个基因进行GO富集分析,未有通路被显著富集,但结果显示,与其他K.marxianus相比,FIM1中NCE101、YPT31、KSH1共3个基因参与了蛋白分泌过程(GO: 0009306),该结果提示FIM1在蛋白分泌领域可能具有良好的应用潜力。
根据同源基因家族分析结果初步构建了12株酵母的泛基因组(表6)。结果显示12株酵母共有的同源基因家族有4 009个,在12个基因组中平均占比为79.88%,附属基因占比20.12%。附属基因中,12株酵母多拷贝核心基因(2 733个)的平均占比为4.54%,其他非必需基因(9 380个)的平均占比为15.58%。在12株酵母中,100656-19含有最多的附属基因,共有2 180个附属基因,其中多拷贝核心基因741个,其他非必需基因1 439个;IIPE453的附属基因最少,共有790个附属基因,其中多拷贝核心基因221个,其他非必需基因569个(表6)。
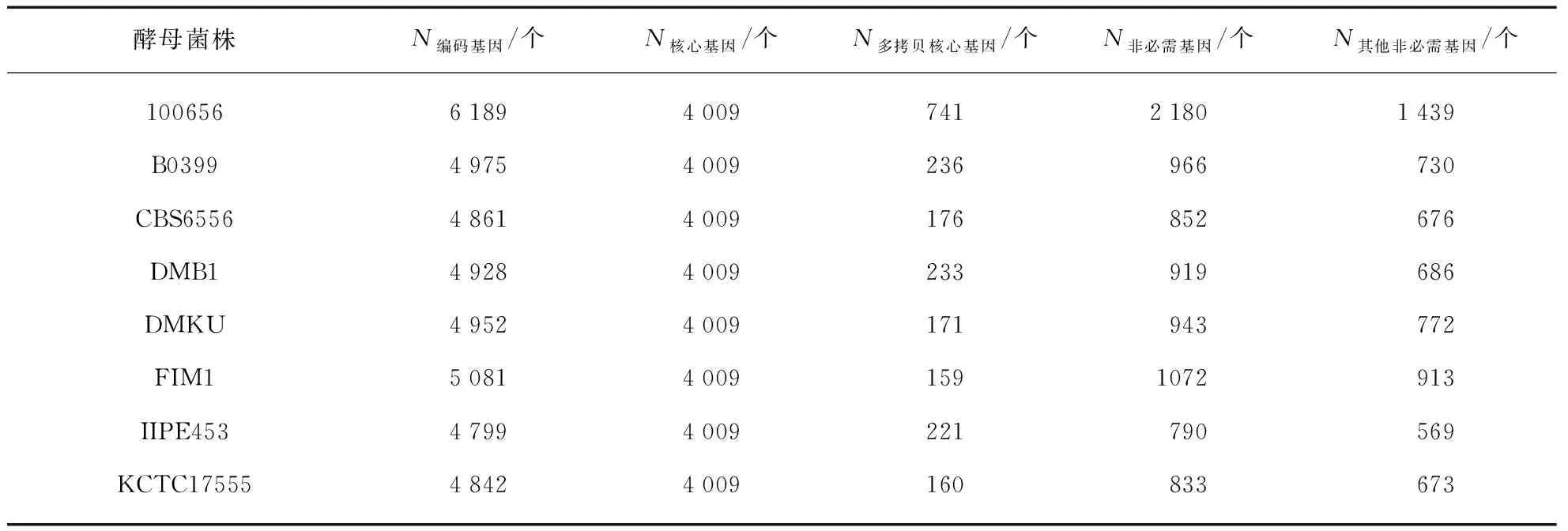
表6 K.marxianus泛基因组分析Tab.6 Pan-genome analysis of K.marxianus
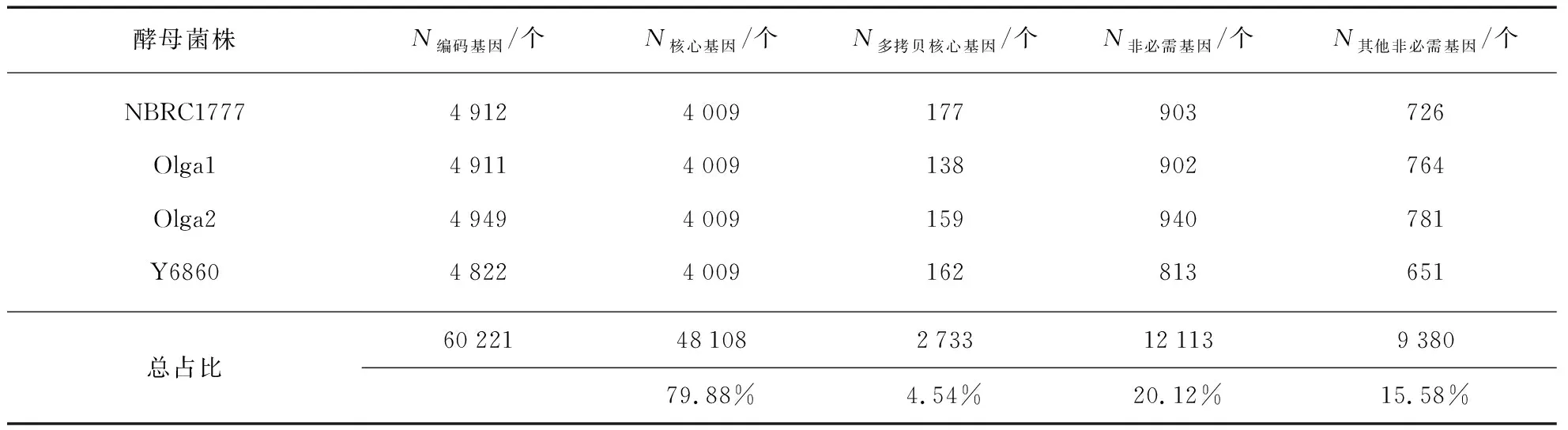
(续表)
基于同源基因家族构建结果,使用PanGP计算了新同源基因家族(N)与基因组数目(G)之间的关系(图5(a)),计算出拟合方程N=1 734.8G-2.77(R2=0.816 7),当K.marxianus基因组数目增加到12时,只有2个新的同源基因家族,同源基因家族总数趋于稳定。随后使用PanGP软件计算泛基因数、核心基因数和基因组数目之间的关系(图5(b))。该软件将泛基因定义为所有同源基因家族的集合,据此拟合得到泛基因的个数(P)与基因组数目(G)关系的为P=128 270G0-123 445(R2=1,精确到小数点后4位),根据方程预测K.marxianus泛基因组中的基因数为4 825个。核心基因的个数(C)与基因组数目(G)关系的拟合方程为C=1 113.91e-0.12G+3 760.1(R2=0.980 9)。根据方程预测K.marxianus的基因组中包含3 760个核心基因。根据新同源基因家族(N)与基因组数目(G)的拟合方程,预测K.marxianus基因组总数增加到15个以上时,不会有新的同源基因家族出现,则泛基因数不会再增加。因此本研究推测K.marxianus的泛基因组是闭合的,泛基因组由4 825个基因组成,核心基因有3 760个。
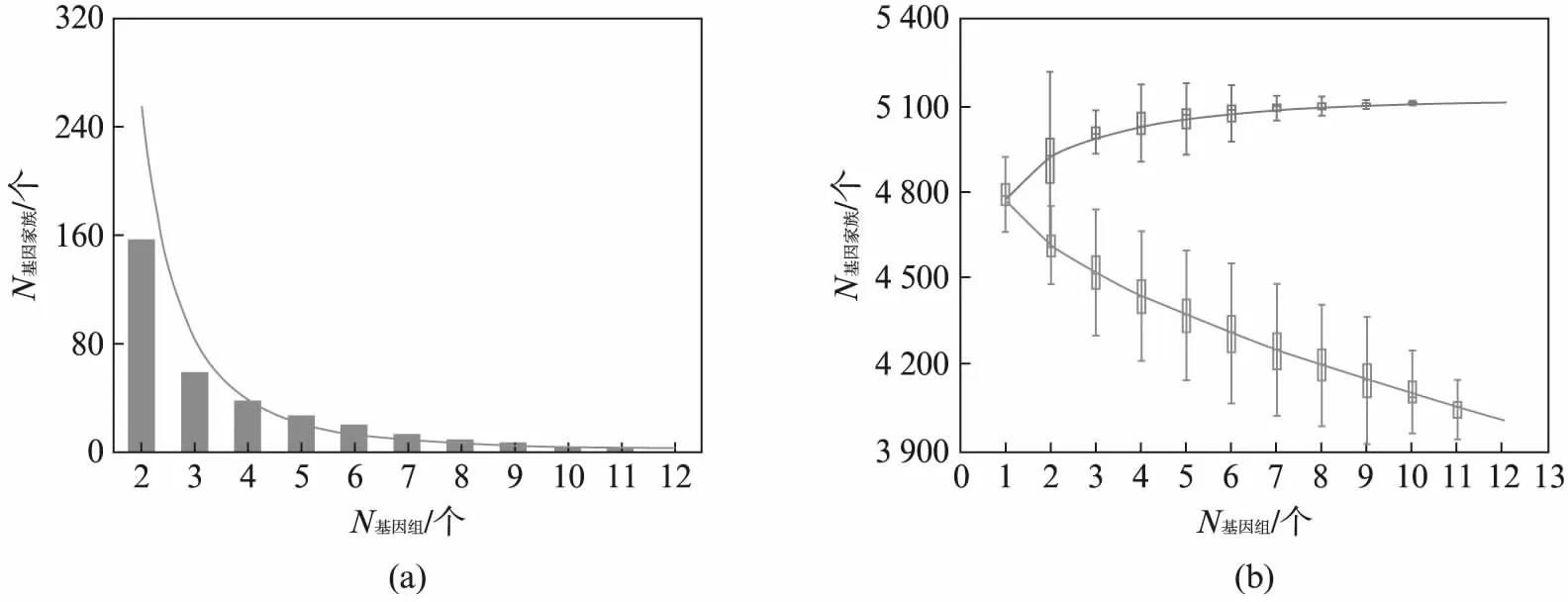
图5 K.marxianus泛基因组分析Fig.5 Pan-genome analysis of K.marxianus(a) K.marxianus基因组数目与新基因家族数目的关系;(b) K.marxianus泛基因组分析,蓝色曲线为泛基因拟合曲线,绿色为核心基因拟合曲线。
3 讨 论
随着测序技术的不断进步,PacBio HiFi测序模式的面世后,3代测序已经可以同时实现重复序列的有效覆盖与测序单碱基的高准确度[27]。这极大地提高了新物种或个体的基因组测序与组装准确度。与此同时,我们也需要对已完成组装的基因组序列进行校验更新,避免它们被淘汰。本文首先使用重测序的DNA-seq数据校验了FIM1基因组序列,随后对校验前后FIM1的基因组质量进行了评估。BUSCO评估显示校验后FIM1的基因组质量未发生改变,查询默认参数后发现,BUSCO默认blastp的E-value值为1.0×10-3,比对阈值宽松,所以删减基因中的短序列片段可能不会影响评估结果[9]。随后,研究以有序列的删除的HHT1(Histone H3)基因为例,对校验结果进行再评估(图2)。结果显示校验提高了HHT1基因的序列精度,说明DNA-seq数据校验有助于提高FIM1基因组信息的质量。在未来的研究中,获得新的基因组测序数据后,可以将新的数据用于校验已完成组装的基因组序列,从而不断提高序列的精度。不仅是基因组序列,基因注释也不可能一蹴而就。随着各种注释方法与基因功能数据库的不断完善,我们同样需要持续补充与优化注释,这样才能更好的将基因组信息用于实验研究中。
随后,本文构建了K.marxianus泛基因组,拟合方程后预测核心基因数为3 760个左右,K.marxianus泛基因组是闭合的,菌株间基因数目较稳定,较少存在基因水平转移(Horizontal Gene Transfer, HGT)的物种间基因交流现象[28]。HGT在原核生物中是一种重要的增加遗传多样性的方法[29],但在现有报道中,原核生物很少能直接通过HGT事件将基因转移到真核生物基因组中[30]。McCarthy分析了真核生物核心基因与附属基因的起源后认为,对于真核生物,能体现遗传多样性的非必需基因,主要通过基因复制或者基因收缩/扩增现象产生,而不是HGT[22]。结合本文结果,可推测K.marxianus主要通过基因组内部的片段重排、基因复制或者基因收缩扩增的方式,增加自身的遗传多样性,提高进化速率。
最后,NCBI现有的注释预测K.marxianus基因的数目在4 912~5 081之间[4-6],均大于本研究预测的泛基因数4 825。该结果提示我们,现有K.marxianus注释中可能存在较多的多拷贝核心基因以及其他非必需基因。所以下一步,我们可以从其他的非必需基因着手,通过实验与信息学分析结合的方式,健全假基因的注释,鉴定它们的功能。基因组信息的分析与研究,这始终是我们推动K.marxianus遗传学研究深入、遗传工程改造加快的不可或缺的一环。
