化学方法加速疏水缔合聚合物的溶解*
2022-07-04罗平亚叶仲斌肖秀婵
李 玺,罗平亚,叶仲斌,4,舒 政,张 健,肖秀婵
(1.成都工业学院材料与环境工程学院,四川 成都 611730;2.西南石油大学化学化工学院,四川 成都 610500;3.西南石油大学油气藏地质及开发工程国家重点实验室,四川 成都 610500;4.成都工业学院,四川 成都 611730;5.海洋石油高效开发国家重点实验室,北京 100028)
0 前言
疏水缔合聚合物是一种在传统的水溶性聚合物的主链上引入少量疏水基团的新型水溶性聚合物[1]。疏水缔合聚合物的疏水基团通过缔合在水溶液中形成动态物理交联网络结构。与部分水解聚丙烯酰胺(HPAM)相比,疏水缔合聚合物具有更高效的增黏能力和优异的剪切稀释性,还能克服HPAM 不抗盐的弱点。此外,良好的耐温和黏弹性使疏水缔合聚合物在高温油藏也有着优异的表现。然而疏水基团的引入导致疏水缔合聚合物溶解速度降低,制约其在油田的推广应用[2-3]。
中国海油和西南石油大学通过国家863计划进行了加速疏水缔合聚合物溶解的攻关研究。例如:改变搅拌器类型和搅拌方式加速溶解熵变[4],利用筛网切割或胶体研磨的方式提高溶胀胶团比表面积[5-6]。截至目前,加速缔合聚合物溶解的研究主要集中在物理方法上。物理速溶方法能够缩短缔合聚合物的溶解时间,但也造成聚合物不可逆的机械降解,严重影响缔合聚合物的增黏能力。也有研究者尝试用提高溶解温度的方法缩短疏水缔合聚合物的溶解时间[7]。但该方法同样造成不可逆的热氧降解,导致聚合物增黏能力大幅降低。现有的有助于加速溶解的办法都因为物理降解直接损害缔合聚合物的抗温、抗盐、高效增黏等效能而无法应用。因此如何加快缔合聚合物的溶解速度成为当前油田化学工作液发展亟待解决的一个重大技术难题[3,8]。
缔合聚合物的松弛时间高于常规聚合物的原因就是其额外的疏水基团之间的缔合作用。如果在溶解过程中可以屏蔽这种相互作用,缔合聚合物的溶解性能就有可能与常规聚合物相当。环糊精(CD)是环状低聚糖分子,呈截顶空心的圆锥体结构[9]。α-CD、β-CD和γ-CD是最常见的环糊精,分别含6、7 和8 个吡喃糖单元。分子外壁亲水、内腔疏水并且能够作为主体包结各种有机分子或离子客体形成包结络合物,在医药、食品、化学分析、农业及其他工业都有广泛的应用[9-11]。环糊精与疏水基团有极强的亲和性,能够包合缔合聚合物上的疏水基团,拆散缔合聚合物溶液中的缔合结构,大幅降低溶液黏度[11-13]。
本文利用环糊精对疏水基团的包合特性,在缔合聚合物溶解时向溶剂中加入环糊精,以期加速疏水缔合聚合物的溶解过程。环糊精对疏水基团的包合屏蔽了疏水基团本来的缔合作用,导致溶液网络结构的破坏,需要对含有环糊精的缔合聚合物溶液的流变性能进行调整(恢复)。因此,又利用环糊精对客体的竞争包合特性,通过向溶液中加入比缔合聚合物中的疏水基团亲和性更强的客体分子,重新恢复溶液的流变性能。最终实现既缩短缔合聚合物的溶解时间又避免影响其高效增黏效率的目的。
1 实验部分
1.1 材料与仪器
部分水解聚丙烯酰胺(3630S),法国爱森絮凝剂有限公司;疏水缔合聚合物(AP-P4),四川光亚聚合物化工有限公司;α-CD、β-CD、γ-CD,成都市科龙化工试剂厂;模拟注入水,矿化度9158.19 mg/L,离子组成(单位mg/L)为:Na++K+2514.33、Ca2+568.90、Mg2+381.34、HCO3-190.62、SO42-36.64、Cl-5466.36。聚合物的主要理化性能指标见表1,环糊精和疏水缔合聚合物的分子结构见图1。

图1 β-环糊精(a)和疏水缔合聚合物(b)的分子结构
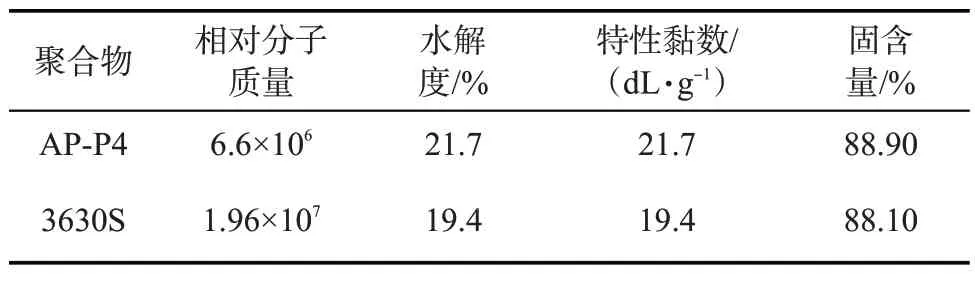
表1 聚合物的理化参数
数显悬吊臂搅拌器,江苏亿晨搅拌科技有限公司;DV-Ⅲ黏度计,美国博勒飞公司;Haake MARSⅢ旋转流变仪,德国赛默飞世尔公司;Nikon SMZ1500 光学显微镜,日本尼康公司;电子天平,量程120 g,精度0.0001 g,上海精密科学仪器有限公司。
1.2 实验方法
(1)溶液流变性能测试。温度为25 ℃时,用黏度计测定溶液在剪切速率为7.34 s-1条件下的表观黏度;用旋转流变仪,在线性黏弹区0.1~10 Hz内进行动态频率扫描,测定储能模量(G')和耗能模量(G'')。
(2)溶解时间的测定。在25 ℃下,溶解聚合物和适量环糊精于模拟水中,配制5 g/L 的聚合物溶液。固定搅拌器转速300 r/min,每隔5 min 测量溶液的表观黏度,直至多次测定得到的溶液黏度值平稳。以溶解过程中聚合物溶液黏度变化率小于5%的溶解用时为聚合物的溶解时间。
(3)溶胀时间的测定。在25 ℃下,用光学显微镜观察聚合物在模拟水或含有不同浓度β-CD 的模拟水中的溶胀过程。选取相近粒径(0.63~0.85 mm,40~20 目)的聚合物干粉颗粒放入溶剂中,启动连续摄像系统并开始计时。以干粉颗粒全部透明作为计时终点,多次重复实验取平均值,以此时间为聚合物的溶胀时间。
2 结果与讨论
2.1 聚丙烯酰胺和疏水缔合聚合物的溶解
将5 g聚合物干粉溶于1 L溶剂的过程中,两种类型聚合物溶液的黏度随时间的变化见图2。在溶解过程中,聚合物分子既要移动大分子链的重心又要克服链间的相互作用,因此扩散很慢。相对地,小分子量的溶剂分子能迅速进入聚合物内部。聚合物的常规溶解过程通常分两步进行。首先小分子量的溶剂分子渗入聚合物的自由体积和空隙中,聚合物溶胀;而后聚合物链段解缠结(chain disentanglement),从溶胀胶团中脱离并均匀分散在溶剂中[14-16]。从图2可以看出,分子量略小的AP-P4的溶解时间比3630S 长得多,且有一个较长时间的黏度平稳期,而3630S 溶解时溶液黏度的增加较为迅速。这是由于两种聚合物在解缠结阶段所需克服的阻力不同。3630S溶解时仅需克服分子间的范德华力,而AP-P4 解缠结还需额外克服疏水基团之间的缔合作用[1,6]。因此,AP-P4 需要溶胀更长时间,使分子间距离增大,分子间缔合作用下降后,才能开始溶解。
教师引导学生思考:为什么会出现不同的结果?学生推测,当R型菌携带大量S型菌荚膜多糖时,才可能发生回复突变。接着,针对如何验证该推测是否正确,教师引入书本上第三组实验:将加热杀死的S-Ⅲ型细菌注射入小鼠体内,小鼠不死亡。证明无R型菌存在下,单独的S型菌荚膜多糖无致死效应。因此格里菲斯做了第四组实验,将R-Ⅱ活菌+杀死S-Ⅲ菌混合物(荚膜多糖)注入小鼠体内。结果发现:小鼠死亡,在死亡的小鼠体内分离出S-Ⅲ活菌,并可稳定遗传。这说明格里菲斯的推测是正确的,由此引入“转化”的概念。教师引导学生提出解释实验结果的假说:加热杀死的S-Ⅲ型菌中含有“转化因子”,使R-Ⅱ型活菌转化为S-Ⅲ型活菌,
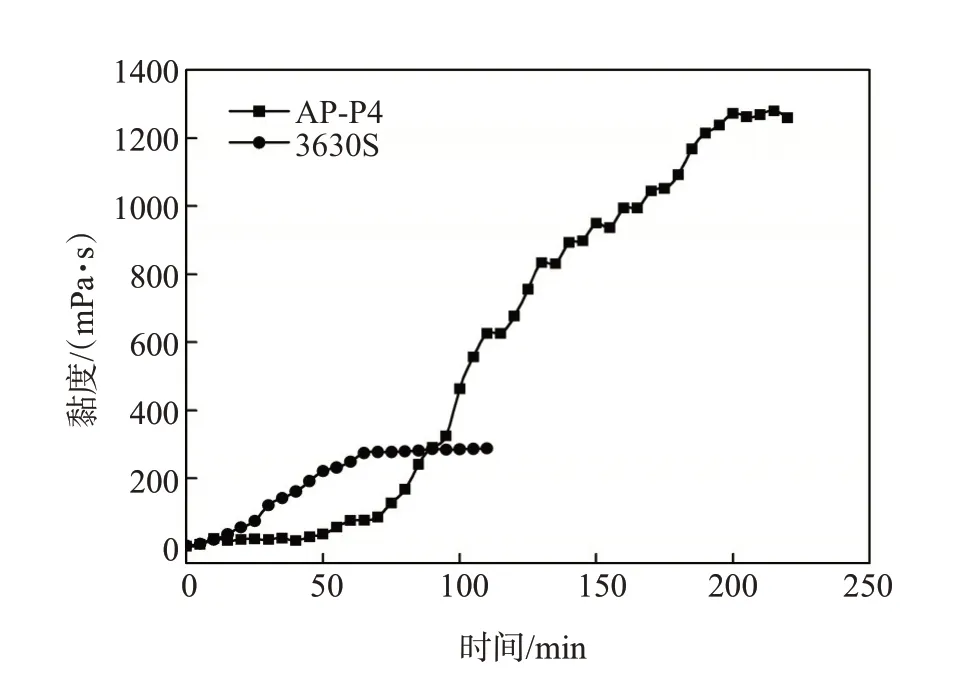
图2 AP-P4和3630S溶液黏度随时间的变化
2.2 环糊精对疏水缔合聚合物溶解的影响
在含有不同环糊精和疏水基团物质的量之比(CD∶[H])的模拟水中,测定了聚合物溶液黏度随溶解时间的变化。由于不同环糊精加量的聚合物溶液黏度降低程度不同,为了便于比较,定义不同时间溶液表观黏度与最大表观黏度的比值为相对表观黏度。不同β-CD 加量条件下溶液的相对表观黏度随时间的变化见图3。AP-P4 的溶解时间和黏度平稳期均随CD∶[H]的增加而明显缩短。当β-CD∶[H]≥4时,AP-P4溶解时间的缩短相对缓慢,且几乎观察不到黏度平稳期,表现出与常规HPAM类似的行为。在α-CD 和γ-CD 存在下,溶液相对黏度与溶解时间的关系曲线与含有β-CD的情况类似,聚合物溶解时间和黏度平稳期都大幅缩短。
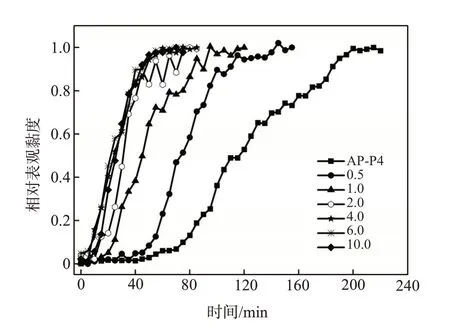
图3 环糊精和疏水基团物质的量比对AP-P4溶液相对黏度与时间关系的影响
在CD∶[H]不同的条件下,α-CD、β-CD、γ-CD对AP-P4溶解时间的影响见图4。随CD∶[H]的增加,溶解时间逐渐降低。当CD∶[H]=10时,AP-P4在含有α-CD、β-CD、γ-CD的模拟水中的溶解时间分别为45、50、60 min,与3630S相当(65 min)。水分子溶剂化疏水基团导致自身分子簇结构被破坏,体系焓增加。环糊精部分替代水分子溶剂化疏水基团,减少了水分子簇结构的破坏,降低了溶解混合热(△HM)[17]。环糊精包合的疏水基团越多,混合热降低得越多,△HM值愈小,自发溶解的倾向愈大[15,18]。
由3 种环糊精对AP-P4 溶解时间的影响(图4)可见,α-CD 具有最强的加速溶解效果,β-CD 其次。这和环糊精疏水空腔的大小与疏水基团尺寸的匹配性相关。AP-P4疏水烷基链的直径小于0.49 nm,α-CD 疏水空腔直径为0.47~0.53 nm,β-CD 和γ-CD依次增大[12,19]。因此,α-CD疏水空腔与疏水烷基链的结合最紧密,其加速溶解效果最显著。但β-CD的价格最低,已经大量工业化生产,如果综合考虑原料成本、效果等因素,利用β-CD 加速缔合聚合物的溶解可能最具应用前景。
图4 环糊精和疏水基团物质的量比对AP-P4溶解时间的影响
为了定量分析CD∶[H]对AP-P4溶解时间的影响,对图4 中CD∶[H]与溶解时间的数据进行了拟合。由于零不能取对数,以一个极小值0.001 来代替不含CD 的模拟水中AP-P4 的溶解时间,其物理意义为加入极少量的环糊精(CD∶[H]=0.001)对AP-P4的溶解时间无影响。由拟合结果(表2)可见,3 条曲线都较好地符合指数关系,溶解时间随CD∶[H]的增加呈指数递减。

表2 环糊精和疏水基团物质的量比与AP-P4溶解时间的拟合关系
2.3 环糊精对疏水缔合聚合物溶胀的影响
疏水缔合聚合物固体颗粒在溶胀过程中,水分子不断渗透,形成透明胶团,未渗透部分不断缩小直至消失。在含有不同浓度β-CD的溶液中,多次测量AP-P4颗粒的溶胀时间见表3。在β-CD含量分别为0、0.1%、0.5%的模拟水中,AP-P4 颗粒的平均溶胀时间分别为461、480、425 s。由于在不同溶剂中,平均溶胀时间变化不明显,而在相同溶剂中,AP-P4颗粒溶胀时间的波动较大,表明溶胀过程受环糊精含量影响较小,波动较大主要与聚合物干粉颗粒的微小空隙和自由体积大小有关[9]。比较溶解时间和溶胀时间长度,发现溶胀过程仅仅占整个溶解过程的很小一部分,大量的时间都用于聚合物链段的解缠结过程。

表3 AP-P4在β-CD加量不同的模拟水中的溶胀时间
从黏度随时间变化的曲线(图2 和图3)可以看出,AP-P4 的黏度平稳期持续约60 min,且随CD∶[H]的增加,AP-P4 黏度平稳期迅速缩短。这可能是由于缔合聚合物溶胀形成的凝胶层中的聚合物链段在分子间相互作用和强烈的缔合作用下结合得比较紧密,难以解缠结从凝胶层进入溶液中,溶液黏度增加较慢[14,20-21]。当CD∶[H]≥4时,疏水基团被环糊精大量包合,缔合作用被屏蔽,聚合物链段更容易解缠结,因此溶液黏度平稳期消失,表现出与聚丙烯酰胺类似的黏度-时间变化行为。
2.4 环糊精对疏水缔合聚合物溶液流变性的影响
环糊精对疏水基团的包合能明显加速疏水缔合聚合物的溶解过程,但也因此对缔合聚合物溶液的流变性质有较为强烈的影响[12-13,22]。在加量为0.5%的条件下,α-CD、β-CD、γ-CD 对缔合聚合物表观黏度的影响见图5。缔合聚合物溶液表观黏度随CD∶[H]的增加而迅速降低。当CD∶[H]≥4 时,溶液黏度基本稳定。疏水缔合聚合物溶液黏度由结构黏度和非结构黏度构成[17]。环糊精包合疏水基团后,疏水缔合作用被屏蔽,导致溶液结构被破坏,结构黏度大幅度降低。溶液的整体黏度也因此而大幅降低。当CD∶[H]≥4时,溶液黏度降低不再明显,表明分子间的疏水缔合已大体消除,此时溶液黏度主要由非结构黏度构成。在0.5%的AP-P4 溶液中,结构黏度占比超过97%,而非结构黏度不到3%。这也表明,与分子量相当的常规聚丙烯酰胺相比,缔合聚合物能通过疏水基团形成缔合结构大幅提高溶液黏度。

图5 环糊精和疏水基团物质的量比对AP-P4溶液表观黏度的影响
进一步研究了β-CD 对AP-P4 溶液动态模量的影响,结果如图6所示。随CD∶[H]的增加,G'和G''的频率依赖性增加。根据瞬态网络理论[27,12],G'与聚合物溶液网络结构的活性连接点数量呈正比,G'下降反映环糊精屏蔽了缔合作用,聚合物溶液网络结构被破坏。随CD∶[H]的增加,低频区的G'下降约两个数量级,与结构黏度变化的程度一致。
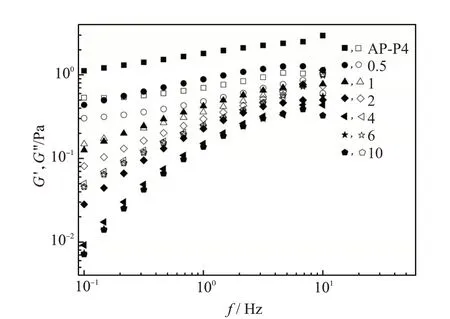
图6 β-CD和疏水基团物质的量比对AP-P4溶液模量的影响
de Gennes 将橡胶状过度到液体状的时间称为最终松弛时间(terminal relaxation time,τ)t。τt也被看作是链解缠结的时间[18],通常用低频下的最长松弛时间(the longest relaxation time,TR)代表τt[23-24]。按式(1)计算TR:

其中,ω—角频率,rad/s;G′—储能模量,Pa;G′′—耗能模量,Pa。
0.5 %缔合聚合物溶液的TR随CD∶[H]的变化如图7 所示。TR随环糊精的加入迅速下降,链解缠结时间大幅缩短。继续增加CD∶[H],TR的变化较小。当CD∶[H]=10时,α-CD、β-CD、γ-CD将缔合聚合物溶液的TR从3.34 s 降低了一个数量级,分别缩短至0.25、0.36、0.50 s。根据标度理论,加入环糊精后聚合物溶胀形成的浓稠体系中高分子链的解缠结时间都缩短。因此,环糊精通过包合疏水基团,拆散缔合结构,缩短聚合物链段解缠结时间,实现缔合聚合物的加速溶解。

图7 环糊精和疏水基团物质的量比对TR的影响
2.5 疏水缔合聚合物溶液流变性能的恢复
环糊精包合疏水基团对缔合聚合物的缔合结构造成破坏,导致其表观黏度和动态模量均大幅降低。因此,在使用按这种方式溶解好的缔合聚合物溶液前需要对其流变性能进行调节。可以通过水解环糊精释放疏水基团实现含有环糊精的缔合聚合物溶液黏度的恢复[12,25]。本研究则采用另一种方法,利用环糊精的竞争包合特性,通过加入与环糊精亲和性更强的客体将之前包合物中的客体分子置换出来。向含有环糊精的缔合聚合物溶液中加入具有不同亲水亲油平衡(HLB)值的壬基酚聚氧乙烯醚(NPe,e代表分子平均的乙氧基数),以恢复缔合聚合物溶液的流变性质。由于现场注入地层的AP-P4溶液质量分数通常都低于0.2%,且当CD∶[H]=1 时,聚合物溶解时间已满足现场对溶解时间的要求,故选取质量分数为0.2%的AP-P4溶液作为研究对象,其中CD∶[H]=1。
当β-CD∶[H]=1 时,含不同NPe的0.2%缔合聚合物溶液在7.34 s-1下的表观黏度见图8。随NPe与环糊精物质的量比(NP∶eCD)增加,溶液黏度都呈现先增加后下降的趋势。所有NPe均可提高溶液黏度,但不同HLB 值的NPe对溶液黏度的恢复程度大不相同。随HLB值增加,溶液黏度恢复程度逐渐降低。具有最低HLB 值的NP4能完全恢复并进一步提高溶液黏度,NP7仅能完全恢复溶液黏度,具有较高HLB 值的NP10和NP15只能部分恢复溶液黏度。这应该归因于不同HLB值的NPe与环糊精的亲和力和表面活性剂形成的胶束结构不同。HLB值越低,非离子表面活性剂与环糊精的亲和力越强。低HLB值的NP4能将被包合的疏水基团尽可能多地释放出来。加入NP4后体系黏度最高值超过相同浓度的AP-P4 溶液黏度,是因为缔合聚合物与过量非离子表面活性剂形成的胶束进一步作用,形成了更强的网络结构[23-24,26]。

图8 NPe、β-CD物质的量比对AP-P4溶液表观黏度的影响
NP∶4β-CD 对β-CD∶[H]=1 的0.2%AP-P4(记为AP-P4/CD)溶液动态模量的影响见图9。与图8 的结果类似,随NP∶4CD 的增加,模量先增加后下降,而模量的频率依赖性先下降后升高,且模量的最大值超过相同浓度的AP-P4 溶液。说明随NPe的加入,溶液中聚合物链段间的活性连接数量先增加后减少。链段间活性连接数量的增加应该同时归因于被释放的疏水基团的重新缔合以及NP4与AP-P4的相互作用。模量的再次降低对应过量的NP4与缔合聚合物相互作用导致AP-P4溶液的网络结构被破坏。低HLB 值的NP4能在较宽的比例范围内完全恢复溶液流变性质。若仅要求恢复缔合聚合物溶液的流变性,配液对药剂比例的精度要求不高,有利于现场施工。
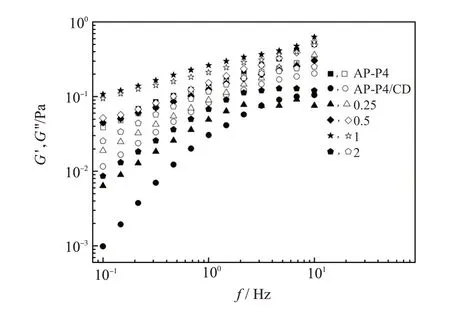
图9 NP4、β-CD物质的量比对AP-P4溶液动态模量的影响
值得注意的是,虽然在NP∶4CD=0.5 时体系的黏度与初始AP-P4 溶液的相当,但动态模量频率曲线并不完全重合,也即模量随频率的变化不完全一致。这表明最终得到的溶液在结构上与初始AP-P4溶液仍然有所差别,也证实了缔合聚合物溶液流变性的变化并非仅仅是由于竞争包合释放出环糊精重新缔合导致,还同时受到缔合聚合物与表面活性剂相互作用的影响。
3 结论
疏水缔合聚合物在溶解过程中,溶胀过程很短而解缠结过程耗时较长。将环糊精加入到溶剂中有助于加速缔合聚合物的溶解。常见的3种环糊精均具有加速缔合聚合物溶解的效果,随环糊精加量增加聚合物溶解时间呈指数下降。受疏水与疏水烷基链的尺寸匹配性影响,α-环糊精的加速溶解效果最强,β-环糊精次之,γ-环糊精最差。综合考虑原料成本、效果等因素,β-环糊精最具工业应用前景。从热力学上看,缔合聚合物溶解的混合热因环糊精的包合而降低,聚合物自发溶解倾向提高。综合分析溶胀、溶解和聚合物流变实验结果,环糊精对缔合聚合物的溶胀过程无明显影响,主要通过包合疏水基团,破坏疏水缔合结构,缩短链段解缠结过程而加速溶解。
环糊精的加入严重降低缔合聚合物溶液的黏度与模量。利用环糊精包合物的竞争包合特性,向含有环糊精的缔合聚合物中加入适量的与环糊精具有更强亲和力的非离子表面活性剂(NPe)可以完全恢复并进一步提高溶液的流变性能。随NPe的HLB值增加,疏水缔合聚合物溶液流变性质恢复程度逐渐降低。本研究采用化学方法既实现了疏水缔合聚合物的加速溶解,又避免了以往速溶方法中物理降解导致溶液流变性能不可逆的损害,有助于疏水缔合聚合物在油田的推广应用。
