多芳基咪唑CPD的合成及与G-四链体相互作用研究
2022-07-01谢晓玲邓南翔
郑 滔, 谢晓玲, 邓南翔, 车 通, 舒 兵*
(1. 广东药科大学 药学院, 广东 广州 510006; 2. 广东药科大学 新药研发中心, 广东 广州 510006)
G-四链体是由DNA链上连续富鸟嘌呤(G)序列形成的一种特殊的核酸二级结构[1].越来越多的证据表明G-四链体参与多种重要的生物学过程,如端粒末端保护、DNA复制、转录和翻译等,并且在细胞增殖、凋亡与衰老、肿瘤的发生及发展过程中都扮演着重要的调控角色[2-4].生物信息学等研究表明,在染色体端粒末端、癌基因的启动子区域以及mRNA的非翻译区域等重要调控区域均存在大量可以形成G-四链体的潜在序列[5].
然而,现阶段对G-四链体的研究只涉及到较少的基因序列,仍有大量G-四链体潜在形成序列有待实验证明[5].而且,G-四链体在细胞内的形成过程、小分子靶向G-四链体之后下调癌基因的表达等方面仍然缺乏最直接的证据[6].这也是目前靶向G-四链体的药物设计与开发一直未得到众多科学家认同的重要原因.因此,开发能够对G-四链体结构进行快速确证的方法和工具显得尤为重要.
多取代咪唑作为发色团主要应用于有机光电二极管、有机光电管、半导体等领域[7].Chen等[8-9]发现在咪唑环的2位引入香豆素或者萘二甲酰胺荧光基团分别得到化合物TSIZ01和IZNP-1(图1),可以增加化合物对G-四链体的荧光识别作用.为获得结构多样性的靶向G-四链体的荧光探针分子,本文拟以多芳基咪唑的结构为基础,通过结构修饰以期得到结构新颖且识别G-四链体的目标化合物.有研究表明,在咪唑的1位引入较大的芳香结构,可以增加化合物对G-四链体的π-π堆积作用和荧光识别作用[10-11].为此,本研究在上述文献的工作基础上,通过在咪唑的1位引入具有芳香荧光性咔唑基团,设计得到新型多芳基咪唑类化合物CPD(图1).采用紫外可见分光光度计测定了CPD的紫外光谱,采用荧光光谱仪和分子对接实验评价了CPD与G-四链体相互作用的荧光性能和相互作用模式.
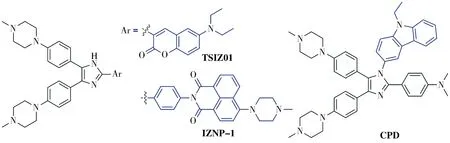
图1 化合物TSIZ01、IZNP-1和CPD的结构
1 实验部分
1.1 主要仪器与试剂DF-101S集热式恒温加热磁力搅拌器(河南巩义宇华有限公司);Fluoromax-4荧光光谱仪(赛默飞世尔科技公司);UV-2600紫外可见分光光度计(赛默飞世尔科技公司);Advance Bruker 400M超导核磁共振波谱仪(瑞士BRUKER公司);Thermo超高效液相色谱串联质谱仪(赛默飞世尔科技公司).4-氟苯甲醛、维生素B1(VB1)、三氯化铁、N-甲基哌嗪、4-二甲氨基苯甲醛、3-氨基-9-乙基-9H-咔唑、NaOH、乙醇、醋酸、醋酸铵等均为分析纯,无需进一步处理.
1.2 实验过程以4-氟苯甲醛为起始原料,在VB1/NaOH催化体系中发生安息香缩合反应得到1,2-双(4-氟苯基)-2-羟基乙酮(2).中间体2经过三氯化铁氧化得到羟基氧化产物2,2-双(4-氟苯基)乙二酮(3).之后3与N-甲基哌嗪发生亲核反应得到2,2-双(4-甲基哌嗪基苯基)乙二酮(4).N-甲基哌嗪的相对于化合物3的量大大过量,主要是为了使得化合物3能够彻底反应完全.最后,中间体4与4-二甲氨基苯甲醛、3-氨基-9-乙基-9H-咔唑在醋酸/醋酸铵体系中发生Debus-Radziszewski咪唑合成反应得到了多芳基取代咪唑CPD(图2).目标化合物的结构经1H NMR、13C NMR和HRMS等方法确证.
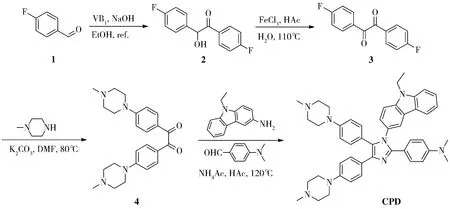
图2 化合物CPD的合成路线
1.2.11,2-双(4-氟苯基)-2-羟基乙酮(2)的合成称取3.6 g(0.01 mmol)维生素B1于250 mL烧瓶中,加入30.0 mL体积浓度95%乙醇,搅拌5 min,滴加5.0 mL质量浓度10% NaOH溶液,搅拌20 min后,滴加10 g(0.08 mol)4-氟苯甲醛,滴加完毕后,保持pH=8~9,回流1 h.反应毕,放冷至室温,旋干溶剂,加入50 mL乙酸乙酯和20 mL水搅拌,分液,水层加乙酸乙酯(20 mL×3)萃取,合并有机相,用饱和食盐水(100 mL×3)洗涤,无水硫酸钠干燥后过滤,滤液减压蒸除溶剂,硅胶柱层析(洗脱剂:V(乙酸乙酯)/V(石油醚)=1/6),共得到1,2-双(4-氟苯基)-2-羟基乙酮(2),白色固体6.2 g,收率62%;1H NMR(400 MHz,CDCl3)δ:8.19~8.07(m,2H),7.47~7.28(m,4H),7.25~7.12(m,2H),6.16(s,1H).
1.2.22,2-双(4-氟苯基)乙二酮(3)的合成 称取5 g(20.16 mmol)中间体2于反应瓶中,加入15 mL冰醋酸,5 mL水,8.6 g(53.0 mmol)三氯化铁,反应液于110 ℃油浴中加热1 h后,停止反应,加入30 mL乙酸乙酯和20 mL水搅拌,分液,水层加乙酸乙酯(3×10 mL)萃取,合并有机层,饱和食盐水(2×30 mL)洗涤,无水硫酸钠干燥,过滤,旋干溶剂,硅胶柱层析(洗脱剂:V(乙酸乙酯)/V(石油醚)=1/32),共得到2,2-双(4-氟苯基)乙二酮(3),白色固体4.1 g,收率83%,1H NMR(400 MHz,CDCl3)δ:7.79~7.71(m,4H),7.53~7.43(m,4H).
1.2.32,2-双(4-甲基哌嗪基苯基)乙二酮(4)的合成 称取3 g(12.19 mmol)中间体3于反应瓶中,加入30 mL DMSO,搅拌溶解后加入2.0 g(14.63 mmol)碳酸钾和2.9 g(29.26 mmol)N-甲基哌嗪,反应液于80 ℃油浴中加热4 h.反应毕,将体系放冷至室温,加入50 ml乙酸乙酯,搅拌5 min后,将混合液倒入200 mL冰水中,搅拌5 min后分液,水层加乙酸乙酯(2×30 mL)萃取,合并有机层,加饱和食盐水(2×30 mL)洗涤,无水硫酸钠干燥,过滤,旋干溶剂,硅胶柱层析(洗脱剂:V(甲醇)/V(二氯甲烷)=1/50,含体积分数0.5%氨水),共得到2,2-双(4-甲基哌嗪基苯基)乙二酮(4),黄色固体3.5 g,收率70%,1H NMR(400 MHz,CDCl3)δ:7.79(d,J=9.0 Hz,4H),6.79(d,J=9.1 Hz,4H),3.41~3.32(m,8H),2.55~2.47(m,8H),2.30(s,6H).
1.2.41-(1-(9-乙基-9H-咔唑-3-基)-4,5-双(4-(4-甲基哌嗪基-1基)-苯基)-2-(4-二甲胺基)苯基-1H-咪唑(CPD)的合成 称取0.5 g(1.23 mmol)中间体4于反应瓶中,加入220.1 mg(1.47 mmol)4-二甲胺基苯甲醛、516.6 mg(2.46 mmol)3-氨基-9-乙基-9H-咔唑和947 mg(12.3 mmol)醋酸铵,加入10 mL冰醋酸,反应体系于120 ℃油浴中加热8h.反应毕,放冷至室温,加入50 mL乙酸乙酯和50 mL冰水搅拌10 min后,滴加饱和碳酸钠溶液调pH=9~10,分液,水层加乙酸乙酯(3×10 mL)萃取,合并有机层,饱和食盐水(2×30 mL)洗涤,无水硫酸钠干燥,过滤,旋干溶剂,硅胶柱层析(洗脱剂:V(甲醇)/V(二氯甲烷)=1/30,含体积分数1%氨水),共得到终产物CPD,黄色固体331.7 mg,收率37%;1H NMR(400 MHz,CDCl3)δ:7.97(d,J=7.7 Hz,1H),7.86(d,J=1.8 Hz,1H),7.57(d,J=8.8 Hz,2H),7.50(t,J=7.6 Hz,1H),7.43(d,J=8.2 Hz,1H),7.33(d,J=8.9 Hz,2H),7.26(s,1H),7.22(t,J=7.4 Hz,1H),7.18(dd,J=8.6 Hz,1H),7.04(d,J=8.7 Hz,2H),6.86(d,J=8.8 Hz,2H),6.67(d,J=8.8 Hz,2H),6.49(d,J=9.0 Hz,2H),4.35(q,J=7.1 Hz,2H),3.23(t,J=9.7 Hz,4H),3.13(t,J=8.8 Hz,4H),2.86(s,6H),2.60(t,J=9.7 Hz,4H),2.51(t,J=8.8 Hz,4H),2.37(s,3H),2.31(s,3H),1.46(t,J=7.2 Hz,3H).13C NMR(126 MHz,CDCl3)δ:149.84,149.80,149.39,147.55,140.46,139.02,137.18,131.96,130.02,129.65,129.45,128.17,126.97,126.36,126.18,122.91,122.62,122.16,120.85,120.77,119.09,119.06,115.71,115.17,111.64,108.75,108.49,55.15,55.03,49.01,48.33,46.13,46.08,40.21,37.79,13.91.HRMS(ESI;m/z).计算值C47H52N8,[M+H]+729.431 5,理论值729.434 1.
1.2.5紫外光谱实验 首先,配制含浓度100 mmol/L KCl、10 mmol/L Tris-HCl、pH为7.4的DNA缓冲溶液buffer.称取定量CPD固体,用DMSO溶解,浓度标定为10.0 mmol/L,于-80 ℃下保存.使用时用buffer稀释.使用紫外光谱仪测定浓度3.0 μmol/LCPD的紫外光谱.
1.2.6荧光光谱实验 配制含有浓度1.0 μmol/LCPD的溶液,以最大紫外吸收波长340 nm为激发波长,以360 nm为发射波长,收集在360~650 nm波长范围内的荧光光谱,然后加入DNA溶液,混匀静置30 s后再继续扫描光谱,得到化合物干预不同DNA的荧光光谱.所用到的DNA序列(5'~3')包括:G-四链体形成序列c-myc(TGGGGAGGGTGGGGAGGGTGGGGAAGG)、c-kit(GGGCGGGCGCGAG-GGAGGGG)、bcl-2(GGGCGGGCGCGGGAGGAAGG-GGGCGGG)、vegf(GGGGCGGGCCGGGGGCGGGG)、k-ras(AGGGCGGTGTGGGAAGAGGGAAGAGGGGGAGG)、pdgf-B(GCGGGGCAGGGAGGGTGGAC-GC)、c-myb(GGGCTGGGCTGGGCGGGG)、RET(GGGGCGGGGCGGGGCGGGGG).双链序列有ds26(CAATCGGATCGAATTCGATCCGATTG);单链序列有ss26(GGTTGGTGTGGTTGG)、dT21(TTTTTTTTT-TTTTTTTTTTTT).所有DNA序列在95 ℃下加热5 min后缓慢冷却退火,其中不同类型的富G序列可以形成不同构象的G-四链体结构.
1.2.7分子对接实验 从蛋白结构数据库(PDB)中获取G-四链体的晶体结构作为受体模型,在进行分子对接之前我们去除单链DNA及非必需结晶水分子.使用Molecular Operating Environment 2019(MOE)分子对接软件对结构进行检查矫正,使用MOE2019自带的程序包进行计算,对获得的30个CPD构象结果进行富集分析,并对CPD与G-四链体的结合力自由能进行计算.
2 结果与讨论
2.1 合成与表征在本合成路线中,将VB1/NaOH催化体系应用于安息香缩合反应的合成方法,相比于传统的氰化物(如氰化钾、氰化钠等)催化法,具有原料易得、毒性很低、便于操作、重现性好、产率高等优点.目前已经较为广泛地应用于有机化学和药物化学的学生实验中.在制备标题化合物的过程中,中间体4与4-二甲氨基苯甲醛、3-氨基-9-乙基-9H-咔唑在醋酸/醋酸铵体系中发生Debus-Radziszewski咪唑合成反应得到标题化合物.多次实验发现,产率较低.为此,主要考察了物料比对反应产率的影响.优化结果显示:当反应条件为:n(4)∶n(4-二甲氨基苯甲醛)∶n(3-氨基-9-乙基-9H-咔唑)∶n(醋酸铵)=1∶1.2∶2.0∶10时,能以较高的产率得到目标化合物.
2.2 化合物CPD与G-四链体相互作用的荧光性能评价利用紫外光谱和荧光光谱研究了CPD与G-四链体DNA及其他类型DNA的相互作用.首先使用紫外可见分光光度计测定了CPD的紫外光谱(图3(a)).发现CPD在340 nm有最大紫外吸收.将1.0当量的DNA分别加入到1.0 μmol/L的CPD溶液中,以340 nm为激发波长,以360 nm为发射波长,收集在360~650 nm波长范围内的荧光光谱,结果如图3(b)~(d)和表1所示.
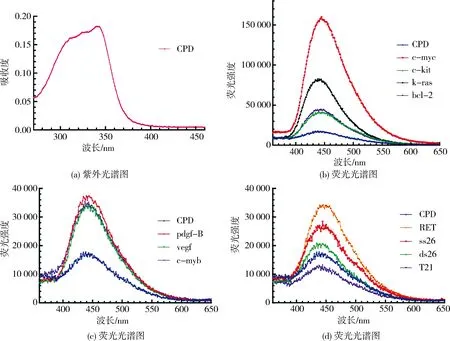
图3 化合物CPD与G-四链体相互作用
在CPD单独存在的情况下,在440 nm处有较弱的发射,荧光响应值为16 950.然而,加入c-myc、c-kit、bcl-2、vegf、k-ras、pdgf-B、c-myb、RET G-四链体后,CPD与G-四链体发生了一定的相互作用,表现在390~550 nm处的荧光发射峰强度发生了一定的变化,并且CPD对不同G-四链体的荧光响应具有一定的差异性.具体表现为:加入bcl-2、c-kit、pdgf-B、vegf、c-myb、RET G-四链体DNA后,CPD在440 nm处的最大发射峰强度略有增加,荧光响应值分别为41 860、41 550、37 320、34 600、33 550、33 460,增幅约为CPD荧光响应强度的2~3倍,可能的原因是CPD与这几类G-四链体发生较弱的相互作用,导致其对这几类G-四链体的荧光响应较差.而加入k-ras和c-myc G-四链体DNA后,同其他G-四链体相比,CPD在440 nm处的最大发射峰强度显著增加,其荧光响应强度分别为80 590(4.8倍)和154 920(9.1倍).尤其是加入c-myc G-四链体,其荧光强度的增幅明显高于其他类似的G-四链体,表现出一定的CPD识别c-myc G-四链体构象的选择性.在同样情况下,单链DNA(T21和ss26)、双链DNA(ds26)均不能使CPD的荧光产生明显的增强(在440 nm处的最大荧光强度分别为12 400、25 980和20 670).以上结果表明化合物CPD可以作为k-ras和c-myc G-四链体的荧光探针,特别是靶向识别c-myc G-四链体.

表 1 CPD与G-四链体的荧光响应强度
2.3 化合物CPD与G-四链体相互作用模式研究为了研究CPD靶向识别G-四链体的作用模式,选择以CPD可以靶向识别的c-myc G-四链体为典型案例,进行了分子对接分析,探讨CPD与G-四链体之间的相互作用模式,结果如图4所示.结果显示CPD的四芳基咪唑骨架较好地堆积在c-myc G-四链体的四分体平面上,与四分体中的鸟嘌呤残基存在π-π相互作用,从而把CPD的咪唑环与荧光基团锁定在一个近平面上.结合能计算结果显示,CPD与c-myc G-四链体的结合自由能达到了-21.3 kcal/mol,表明CPD能很好地与c-myc G-四链体结合.而其他2个苯环结构发生扭曲,使得CPD的两条甲基哌嗪侧链能够伸进G-四链体沟槽内,并与其磷酸骨架的发生较强的静电相互作用.最终导致CPD的分子构象发生变化,因而发射出荧光.
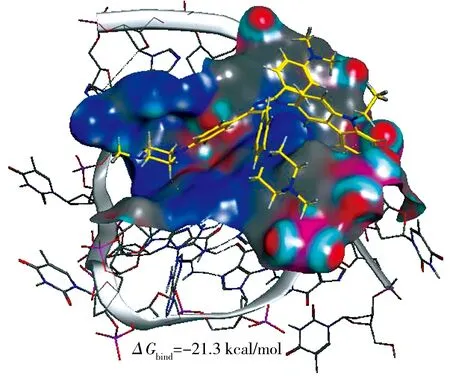
图4 CPD与c-myc G-四链体的分子对接结果(PDB:2l7v)
3 结论
以4-氟苯甲醛为起始原料,通过安息香缩合反应、氧化反应、亲核反应、Debus-Radziszewski咪唑合成反应等步骤合成得到了未见文献报道的四芳基取代咪唑CPD.1H NMR、13C NMR、HRMS等确证其结构.荧光光谱实验研究表明CPD对部分G-四链体具有一定的荧光识别作用,特别是可以识别c-myc G-四链体,并表现出较好的选择性,值得深入研究其作用机制.分子对接结果表明CPD的四芳基咪唑骨架能堆积在G-四分体上,导致原有的分子构象发生变化,因而发射出荧光.在实验过程中发现,由于融合了较强的荧光基团,导致探针分子自身带有一定的荧光响应,这势必影响探针分子的靶向性,值得进一步的结构修饰和优化,为新型靶向G-四链体的荧光探针的发现提供了思路.
致谢广东省中药局科研项目(20212117)和大学生创新创业训练计划项目(202010573010)对本文给予了资助,谨致谢意.
