dMRM 模式下的高效液相色谱串联质谱法同时检测蔬菜中327 种农药残留
2022-07-01李道霞余晓琴周春芳张定秋李澍才
李道霞,周 佳,姚 欢,余晓琴,周春芳,张定秋,李澍才
(四川省食品检验研究院,四川成都 611731)
《中国居民膳食指南》中推荐成人每日蔬菜摄入量为300~500 g[1],蔬菜在日常饮食中所占比重较大。但虫害问题严重影响了蔬菜的质量,农药因具有防治病虫害及调节植物生长的功效,在蔬菜种植过程中发挥着重要作用[2]。然而,长期用药会使害虫和病原微生物产生耐药性,为确保蔬菜质量,通常需要加大用药浓度、增加喷洒次数或多农药联合使用等方式对病虫害进行防治[3]。不合理或不合规的用药方式,导致农药残留问题日益严重,这不仅引起环境污染问题,还严重威胁着消费者的健康[4]。近年来,我国对农药残留问题越来越重视,国家卫生健康委员会不定期地更新农药的最大残留限量标准GB 2763《食品安全国家标准食品中农药最大残留限量》,在2021 年版中对564 种农药的10 092 项最大残留限量进行了严格的规定[5],但农药残留超标现象仍屡禁不止,多农药残留问题也日益突出[6],因此对多农药残留检测技术的要求进一步提高,制定快速、准确、灵敏的多农药残留检测方法具有重要意义。
目前农药残留检测方法主要有气相色谱法(gas chromatography,GC)[7]、液相色谱法(liquid chromatography,LC)[8]、气相色谱-串联质谱(gas chromatography-tandem mass spectrometry,GC-MS/MS)法[9-11]和液相色谱-串联质谱(liquid chromatography-tandem mass spectrometry,LC-MS/MS)法[12-14]等。GC 法和LC 法由于检测器限制,同时检测的农药种类有限,且易受杂质或共存物干扰,准确性相对较差;GC-MS/MS 法虽具有较高的准确度,但在分析较高沸点或低相对分子质量的农药时,易受基质效应、柱流物等因素影响而导致灵敏度较低或产生干扰现象;LC-MS/MS 法灵敏度高,抗干扰能力强,在多农药残留检测中应用十分广泛。目前,LC-MS/MS 法多与QuECHERS 前处理技术联合应用于多农药残留检测[15-17]。dMRM是在多反应监测模式(multi-reactionmonitoring,MRM)基础上以目标化合物的准确保留时间为中心设置采集时间窗,即仪器仅在各目标化合物保留时间前后一段时间内扫描其特征离子对,其优势是根据仪器分析时间对每个化合物的采集时间窗口进行动态分配,仅在设定的时间窗内采集相应的目标组分,这种科学的采集方式可有效减少同时采集的离子对数目,并在一定程度上提高了驻留时间,有效地提高了整个采集时间内的分析效率[12]。
本研究采用QuEChERS 前处理方法结合高效液相色谱-串联质谱法建立了同时测定蔬菜中327 种农药的一种方法,其前处理简单、快速,同时测定的化合物涵盖了有机磷类、有机氯类杀虫剂、三唑类杀菌剂、苯氨基嘧啶类除草剂和杀虫剂等多种农药,使用动态多反应监测模式,提高了分析灵敏度,适用于蔬菜中农药残留的高通量快速筛查和定量检测。
1 材料与方法
1.1 试剂
乙腈、甲醇、甲酸,色谱纯,德国默克公司;乙腈、甲酸、乙酸、无水硫酸镁、氯化钠、柠檬酸钠、柠檬酸二钠、乙酸钠,分析纯,成都市科隆化学品有限公司;实验所用水均为超纯水(GB/T 6682 规定的一级水);N-丙基乙二胺(primary secondary amine,PSA)、石墨化碳黑(graphitized carbon black,GCB)、十八烷基硅烷键合硅胶(C18),Angilent 公司,美国。蔬菜购自成都实体超市。陶瓷均质子,Angilent 公司,美国。
农药混合标准溶液:10 μg/mL,购自天津阿尔塔科技有限公司(货号分别为1ST29392-10B、1ST29766-10B、1ST29394-10B、1ST29386-10T、1ST29395-10A、1ST29387-10B、1ST29390-10A、1ST020032-10A、1ST29551-10M、1ST020414-10A、1ST29398-10A)。
1.2 仪器与设备
配有电喷雾离子源的Angilent LC1290-6460 高效液相色谱-串联质谱仪,Angilent 公司,美国。色谱柱为Waters Acquity UPLC HSS T3(1.8 μm,100 mm×2.1 mm),Waters 公司,美国。漩涡振动器,IKA 公司,德国。Milli-Q高纯水发生器,Millipore 公司,美国。离心机,SIGMA 公司,德国。氮吹仪,中国杭州奥盛科学仪器有限公司;0.22 μm 滤膜,北京泰科瑞迪国际贸易。
1.3 混合标准工作液的配制
精密量取各农药混合标准溶液1.0 mL,氮气挥去溶剂后用乙腈定容至10 mL,配制成各农药标准质量浓度为1 μg/mL 的标准混合溶液,临用新制。
1.4 样品制备
称取5.0 g 均匀样品,置于50 mL 离心管中,精密加入1%甲酸乙腈20 mL,涡旋混匀1 min 后,-20 ℃冷冻10 min,加入QuEchERs 提取包(4 g 无水硫酸镁、1 g 氯化钠、1 g 柠檬酸钠、0.5 g 柠檬酸二钠)和1 颗陶瓷均质子,振荡提取2 min 后超声10 min,6 000 r/min 离心5 min。取上清液7 mL 至含QuEchERs 净化试剂(900 mg 无水硫酸镁、150 mg PSA,含色素较多时采用900 mg 无水硫酸镁、150 mg PSA、15 mg GCB)的离心管中涡旋1 min,6 000 r/min 离心5 min 后精密吸取上清液5 mL,于40 ℃氮吹至近干,精密加入1.0 mL 乙腈溶解残渣,过0.22 μm 滤膜,滤液供液相色谱-串联质谱仪分析。
1.5 仪器条件
1.5.1 色谱条件
色谱柱为Waters Acquity UPLC HSS T3(1.8 μm,100 mm×2.1 mm);流速0.2 mL/min;柱温:40 ℃;进样量2 μL;以0.1%甲酸水为流动相A,0.1%甲酸乙腈为流动相B,线性梯度洗脱,洗脱程序(B%):0~0.5min,10%;0.5~7min,10%~50%;7~21 min,50%~100%;21~23 min,100%,23~23.1 min,100%~10%;23.1~26 min,10%。
1.5.2 质谱条件
离子化模式:电喷雾电离,正负离子模式同时监测(ESI±);质谱监测方式:动态多反应监测模式(dMRM);喷雾电压4 000 V;辅助气气化温度300 ℃;鞘气流速11 L/min;鞘气温度300 ℃;干燥气流速10 L/min。327 种农药的质谱条件具体见表1。
1.6 回收率计算
回收率计算公式见式(1)[18]。

式中,c为加标样品测得浓度,ng/mL;V为样品最终定容体积,mL;n为添加量,ng。
1.7 数据处理
数据处理软件 Angilent MassHunter B.08.01。
2 试验方法建立、条件优化及选择
2.1 dMRM 模式下农药残留检测方法的建立
通过查阅标准、文献和实验室自建农药质谱数据库搜索各目标化合物的特征离子碎片和碰撞能量,通过多反应监测模式采集获得每个化合物的保留时间,针对部分化合物采用手动方式进一步优化质谱参数,根据获得的保留时间等信息建立327 种农药基于动态多反应监测模式的采集方法。该方法通过调整多化合物采集时负载循环时间,以提高灵敏度。dMRM监测模式下100 ng/mL芹菜基质中327 种农药的提取离子流色谱图见图1。

图1 芹菜基质中327 种农药的提取离子流色谱图Fig.1 Extracted ion chromatograms of 327 pesticides in the celery matrix
2.2 前处理条件的优化
以含色素和水分适中的芹菜为基质,以回收率为指标分别对提取溶剂、提取试剂包和净化试剂包的条件进行优化,并将优化后的检测方法应用于豇豆基质。
2.2.1 提取溶剂的选择
本研究的目标化合物包含了有机磷、有机氯、三唑类、拟除虫菊酯类和酰胺类等327 种农药,涉及种类多、极性差异大,因此筛选适合大多数农药的提取溶剂尤为重要。在提取农药时,尽可能地提取出目标物和尽量去除样品的杂质是整个实验的关键,而蔬菜含有大量色素,对农药残留检测影响较大,现有农残类检测标准和文献报道中多采用丙酮[19]、乙腈[20]和1%甲酸乙腈[21]作为提取溶剂,因此本研究考察了丙酮、乙腈和1%甲酸乙腈的提取效果,由图2 可知,当丙酮作为提取溶剂时,提取液中含有较多杂质,对目标物峰型影响校大,这可能是由于丙酮的溶解性很强造成的;乙腈由于其对农药溶解性较强,同时对亲脂性色素等杂质溶解度小,提取液中的色素显著少于丙酮提取液;丙酮提取回收率为0.1%~168.9%,乙腈的提取回收率为0.8%~160.2%,1%甲酸乙腈提取回收率为28.4%~130.9%。试验表明,1%甲酸乙腈的提取效果最好,乙腈次之,丙酮最差。在乙腈中加入甲酸后,93.9%的农药回收率与乙腈差异不大,但2,4-滴丁酸、氟甲喹和磺草唑胺等20 种化合物的回收率显著提高,图3 中列举了两种提取溶剂回收率差异较大的20 种化合物的回收率,因此选择1%甲酸乙腈作为提取溶剂。

图2 三种提取溶剂的整体提取效果Fig.2 Overall extraction effect of three extraction solvents
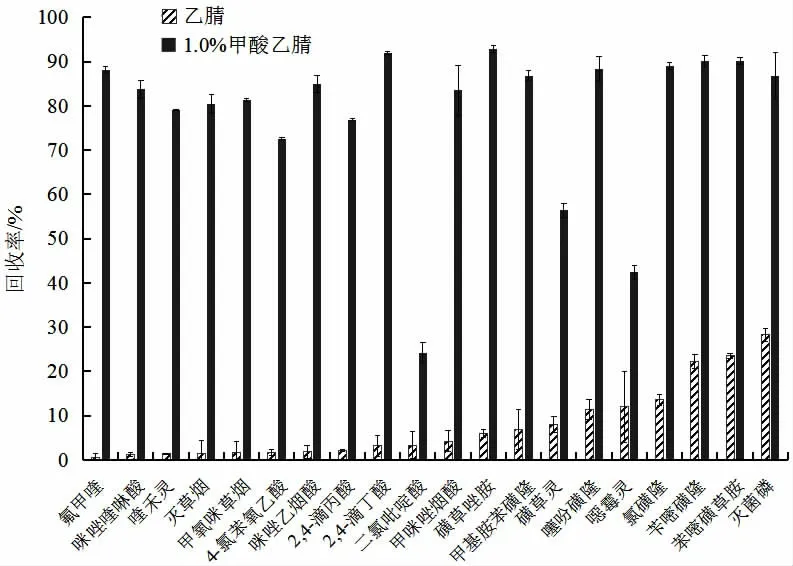
图3 不同溶剂的回收率对比Fig.3 Recoveries of the different solvent extracts
2.2.2 提取试剂包的选择
研究比较了提取试剂Ⅰ(组成为4.0 g 无水硫酸镁、1.0 g 氯化钠、1.0 g 柠檬酸钠和0.5 g 柠檬酸二钠)、提取试剂Ⅱ(组成为6.0 g 无水硫酸镁、1.5 g 乙酸钠)的提取效果,结果显示两种盐包对目标化合物的整体提取效果差异不大,其中回收率在70%~120%之间的化合物占总化合物数量的百分比分别为92.0%和91.1%,如图4 所示,但提取试剂Ⅰ对灭草烟、丁酰肼和十二环吗啉等8 种化合物的提取效果明显优于提取试剂Ⅱ,如图5 所示。综合考虑回收率情况选择提取试剂Ⅰ的组成比例作为提取试剂。
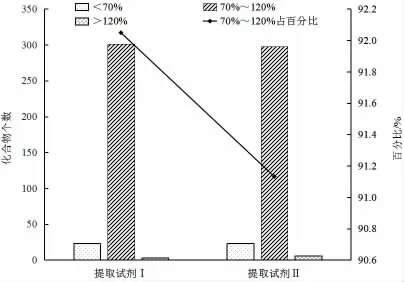
图4 不同提取试剂的影响Fig.4 Effects of different extraction reagents

图5 不同化合物受提取试剂的影响(n=3)Fig.5 Effects of extraction reagents to different compounds(n=3)
2.2.3 净化试剂的选择
蔬菜中含有较多的天然色素,会随着目标化合物一起被提取,净化处理可降低其对目标化合物的干扰。QuEChERS 法中常用的净化材料主要有PSA、C18、GCB和无水硫酸镁等,四种试剂分别具有不同的净化效果[22],因此本研究考察了净化试剂Ⅰ(无水硫酸镁900 mg、PSA 150 mg)、净化试剂Ⅱ(无水硫酸镁900 mg、PSA 150 mg、GCB 15 mg)、净化试剂Ⅲ(无水硫酸镁1200 mg、PSA 400 mg、GCB 200 mg 和C18 400 mg)对芹菜的净化效果,净化后溶液颜色如图6(见下页)所示。
由图6 可知,3 种净化试剂包对目标化合物的整体提取效果具有一定差异,其中回收率在70%~120%之间的化合物占总化合物个数的百分比分别为87.3%(净化试剂Ⅰ)、76.9%(净化试剂Ⅱ)和55.7%(净化试剂Ⅲ),可能是由于GCB 不仅去除叶绿素和极性小分子干扰物的效果较好,对平面结构的目标化合物也有较强的吸附。

图6 不同净化试剂的净化效果Fig.6 Effects of different purification reagents
如图7 所示,拌种胺、涕灭威等12 种化合物回收率受净化试剂包组成的影响较大,随着净化试剂中GCB 含量的增加,试剂的除杂效果越好,但嘧螨醚、嘧菌胺、呋菌胺和噻苯咪唑-5-羟基等10 种化合物的回收率显著降低。综合考虑回收率情况,本研究对于含色素、糖类及脂类较少的基质选择净化试剂Ⅰ,对于含色素、糖类及脂类较多的样品可选择净化试剂Ⅱ。
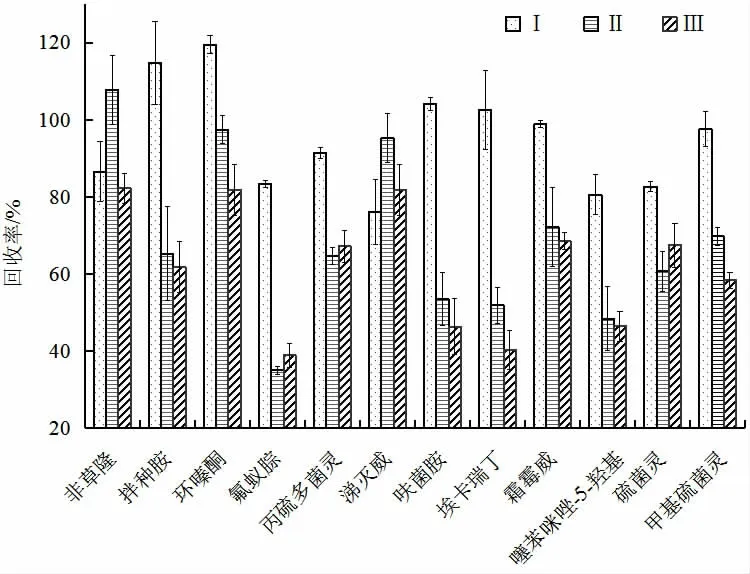
图7 不同净化试剂对回收率的影响(n=3)Fig.7 Recoveries of the different purification reagents (n=3)
2.3 方法学
2.3.1 线性范围与检出限
按1.4 节方法制备芹菜空白基质溶液,用该溶液配制系列质量浓度的基质匹配混合标准曲线(5~300 μg/L),按1.5 节选定的仪器条件进样检测,以各化合物的质量浓度(x,mg/L)为横坐标,定量离子色谱峰面积(y)为纵坐标,考察327 种农药的线性关系。结果显示,各目标化合物在相应的浓度范围内线性关系良好,相关系数(r)均大于0.995。采用空白样品添加低质量浓度的混合标准溶液方式考察327 种农药的检出限(S/N=3)和定量限(S/N=10),检出限范围为0.5~15.0 μg/kg,定量限为1.0~30.0 μg/kg。
2.3.2 准确度和精密度
以空白芹菜为基质,分别制备40、100、200 μg/kg 添加水平的加标样品,按1.4 节进行前处理后进行测定,以考察方法的准确度(以加标回收率表示)和精密度(以相对标准偏差表示)。由表1 可知,方法的准确度为31.7%~133.0%,精密度为0.1%~14.8%,加标回收率在80%~120%之间的化合物的百分比为78.4%。二氧威、二氯吡啶酸、苯醚氰菊酯、醚菊酯、吡蚜酮、哒螨灵、烯禾啶、喹螨醚、环嗪酮、氟胺氰菊酯和磺草灵11 种化合物的回收率均小于60%,经过反复试验其回收率稳定在30%~55%之间。本方法对除上述11 种化合物外的316 种农药具有较高的准确度和精密度,可用于蔬菜中农药残留的高通量快速筛查和确证,对于上述11 种回收率相对较低的化合物采用本方法可进行快速定性筛查。
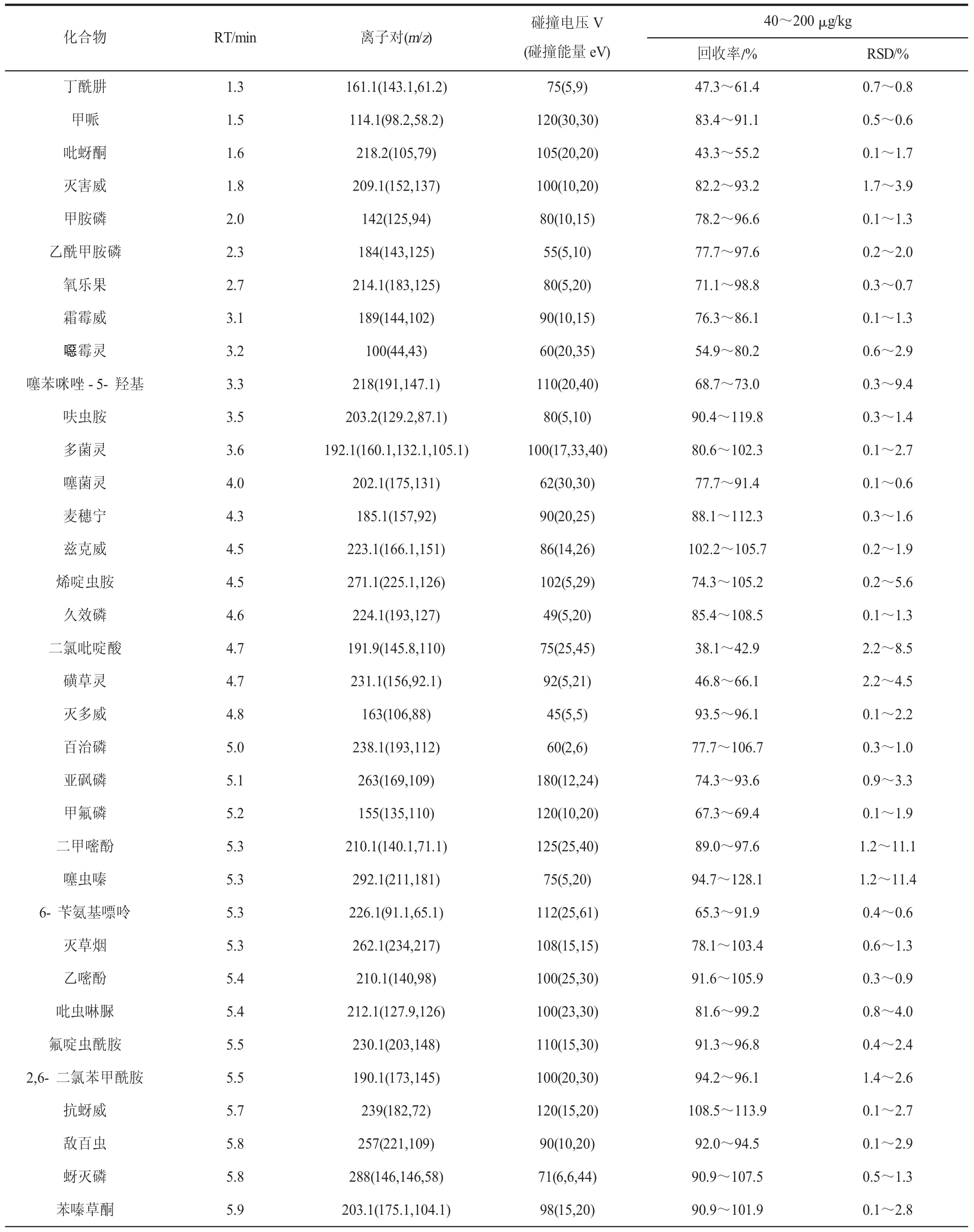
表1 327 种农药的保留时间(RT)、dMRM 参数、加标回收率和相对标准偏差(n=6)Table 1 Parameters of retention times (RT),dMRM,spiked recoveries and RSDs of the 327 pesticides (n=6)

续表

续表
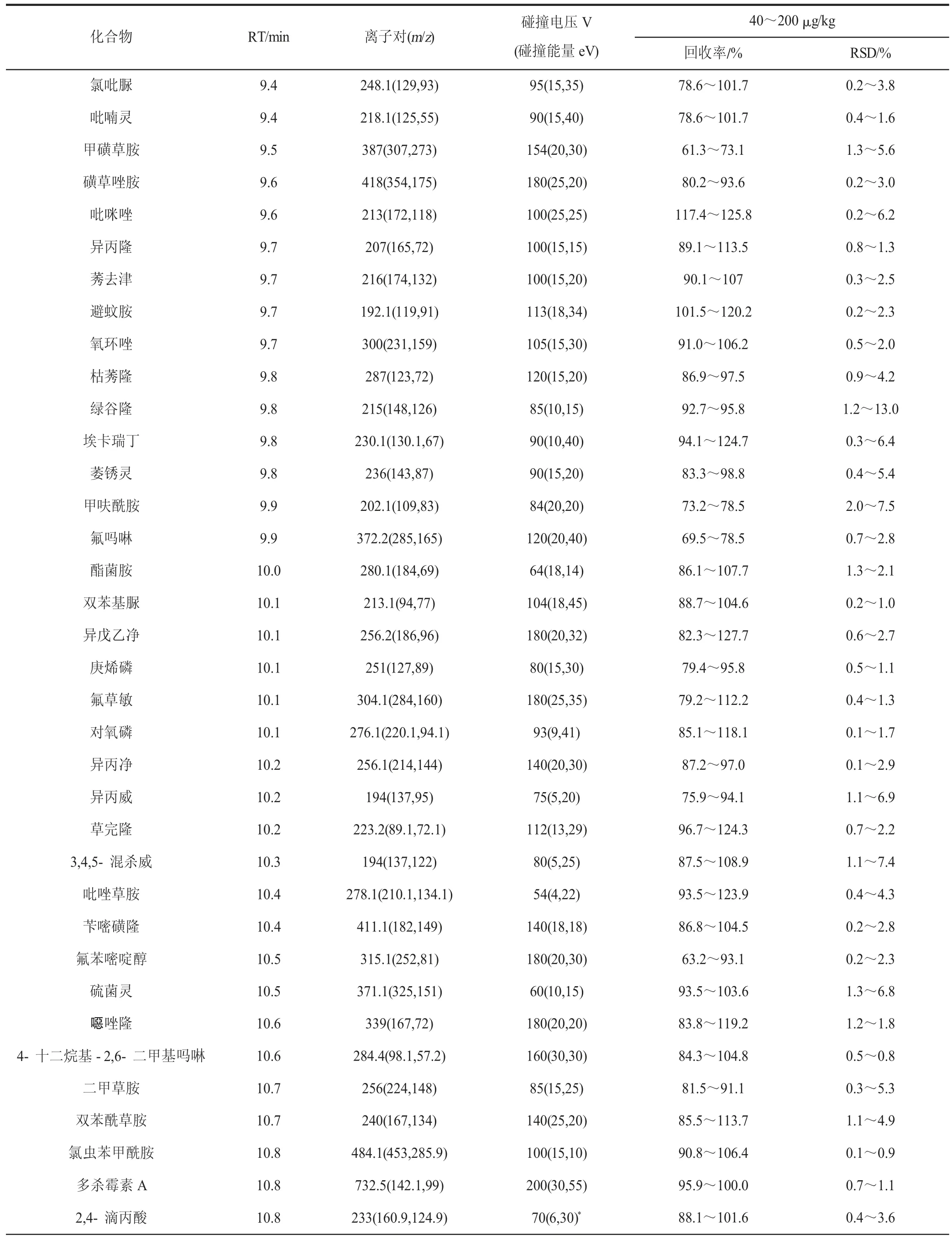
续表

续表
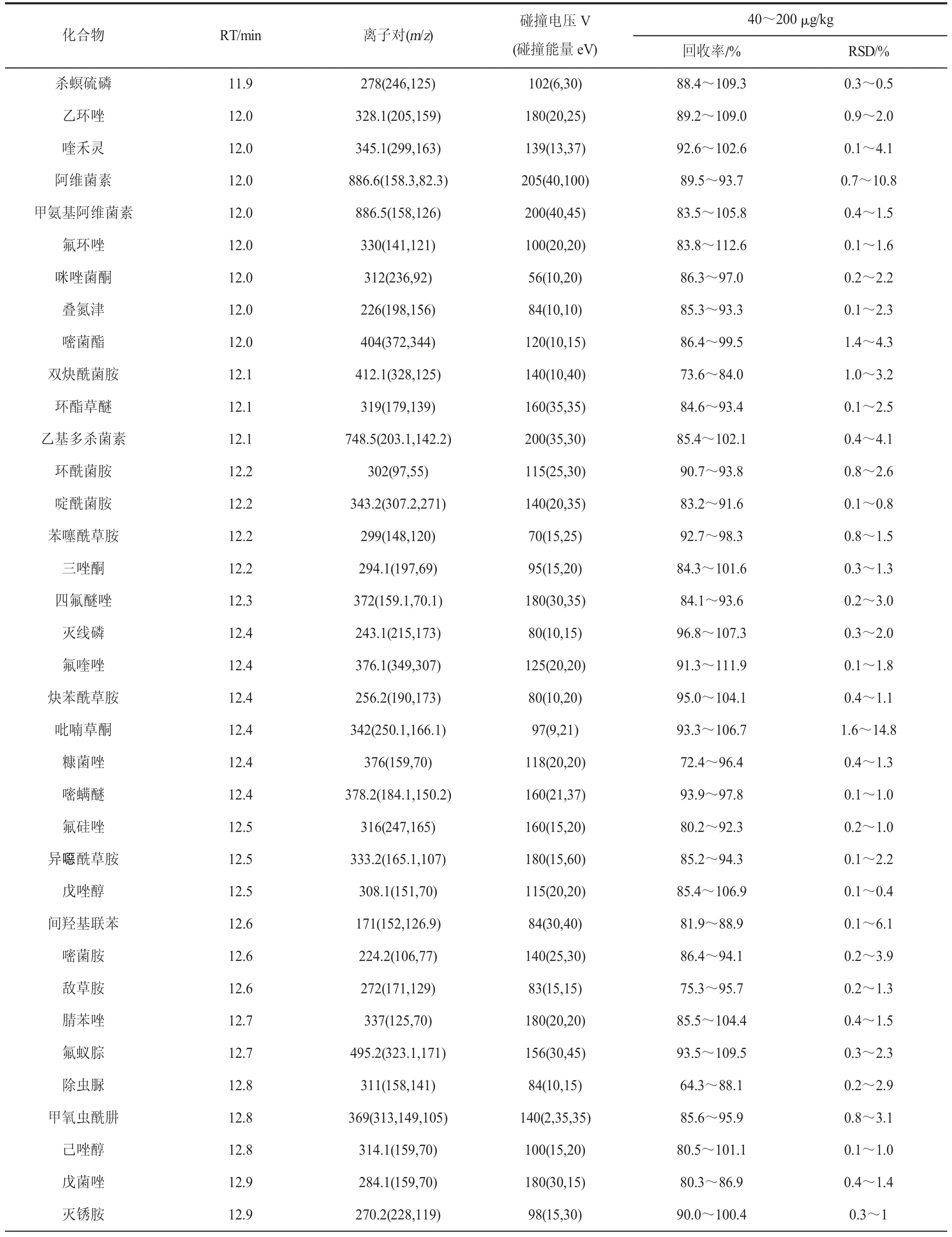
续表

续表

续表

续表

续表
2.3.3 基质效应的考察
基质效应(ME)是离子源中共存组分之间存在离子化竞争引起,可导致目标离子的信号增强或抑制,可采用空白基质匹配标准曲线的斜率与溶剂标准曲线的斜率之比表示,当0.8≤ME≤1.2 时表示弱基质效应,可忽略不计;当0.5≤ME<0.8 或1.2<ME≤1.5 时,表示中等基质效应;当ME<0.5 或ME>1.5 时表示强基质效应;ME<100 时为基质抑制效应,ME>100 时为基质增强效应[23-24]。基质效应考察结果显示57.9%的化合物为基质增强效应,42.1%的为抑制效应;81.4%的农药具有弱基质效应,16.5%具有中等基质效应,仅氯草敏、苯醚甲环唑、氟虫腈、磺草唑胺、磷酸三丁酯、嘧菌酯和噻吩磺隆具有强基质效应(占2.1%),其中氯草敏为抑制效应,其余6个均为增强效应。本实验采用基质匹配标准曲线,可降低基质效应对目标化合物的影响。
2.3.4 方法应用
将建立的检测方法应用至豇豆基质中:以空白豇豆为基质,分别制备40、100、200 μg/kg 添加水平的加标样品,按1.4 节进行前处理,在1.5 节条件对加标样品进行测定,以基质匹配标准曲线法定量,分别考察327 种农药在豇豆基质中的线性范围、相关系数、定量限、准确度、精密度及基质效应等。结果显示,327 种农药在5~300 μg/L 浓度范围内线性关系良好,相关系数(r)均不小于0.991;检出限范围为0.5~15.0 μg/kg,定量限为1.0~30.0 μg/kg;327 种化合物回收率整体分布情况如图8 所示。除丁酰肼、二氯吡啶酸、磺草灵、二氧威、吡喃灵和丁嗪草酮6 种回收率小于50%以外,其余321 种农药在3个加标水平的回收率范围分别为59.1%~139.0%(RSD范围为0.5%~11.8%,40 μg/kg)、60.0%~130.0%(RSD范围为0.4%~9.6%,100 μg/kg)和56.9%~119.5%(RSD范围为0.2%~12.0%,200 μg/kg),丁酰肼、二氯吡啶酸、磺草灵、二氧威、吡喃灵和丁嗪草酮6 种农药的回收率稳定在32.7%~56.5%之间,RSD%均小于9.2%。豇豆基质效应考察结果显示:52.0%的化合物为基质增强效应,47.7%的为抑制效应;73.7%的农药具有弱基质效应,19.6%具有中等基质效应,6.7%具有强基质效应。

图8 豇豆中回收率分布情况Fig.8 Distribution of recoveries in cowpea
3 方法验证
采用建立的方法对10 批经食品安全国家标准或者食品补充检验方法检测为阳性的蔬菜进行检测,全部样品均检出农药,测定结果与标准方法结果一致,结果见表2。
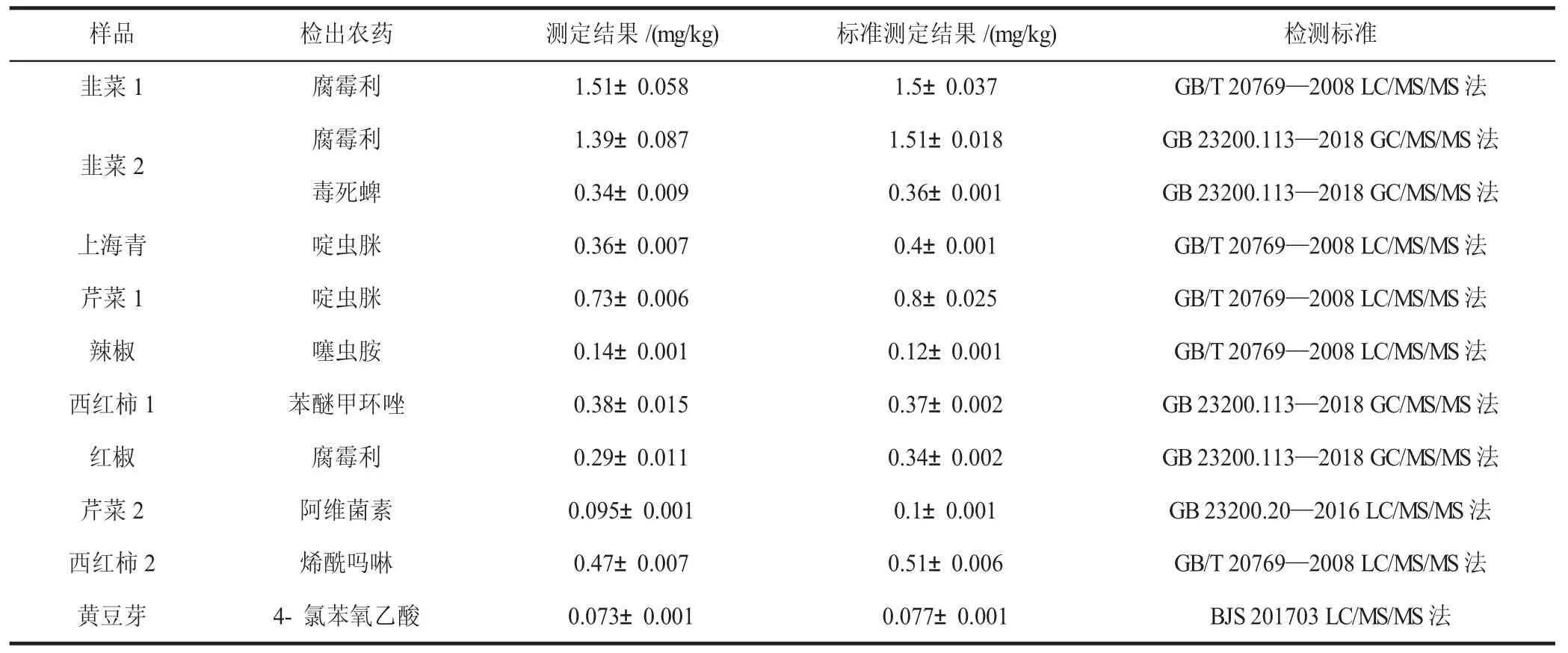
表2 10 批样品农药残留的测定结果Table 2 Determination results of pesticide residues in 10 batches of samples
4 结论
本研究采用高效液相色谱-串联质谱的动态多反应监测模式,建立了蔬菜中327 种农药残留的高通量快速检测方法。通过线性、准确度、精密度、检出限、定量限和实际样品测定,验证了方法的可行性。该方法操作简便、快速、灵敏度较高、准确度和精密度均较好,适用于蔬菜中327 种农药残留的高通量快速筛查和测定。
