4例与GJB2基因相关的遗传性耳聋家系的鉴别诊断
2022-07-01齐科研闫有圣王一鹏阴赪宏
齐科研,杨 锴,闫有圣,王一鹏,阴赪宏
据第二次残疾人抽样调查显示,我国听力残疾者数量达2780万。其中50%以上病例是由遗传学因素所导致的,不合并其他表型的非综合征性耳聋(non-syndromic hearing impairment,NSHI)占70%。我国通过近20年大规模的先天性耳聋分子流行病学调查,逐步建立“孕前-产前-出生后”三级防控体系,为减少聋儿出生或早期干预提供依据。多种分子遗传学检测技术的应用,也极大提高了先天性耳聋患者的检出率。GJB2基因(间隙连接蛋白2,MIM编号*121011,又名Connecxin 26,CX26)编码间隙连接复合物的一个重要亚基,帮助相邻细胞间形成液体孔隙通道,实现细胞间的物质流通。GJB2基因突变是引起非综合征遗传性耳聋最常见的原因,我国有21%中重度以上NSHI患者携带有GJB2基因突变。然而,由于GJB2突变所引发疾病的异质性和表型可变程度很强,需多科室综合分析,以提供准确的咨询意见及再生育指导意见。
本研究纳入了4例先天性耳聋家系,对其进行全面临床表现、家族病史评估,并利用耳聋基因芯片、全外显子组测序技术进行遗传学检测,Sanger测序对特定位点验证,从而明确其发病原因。并且对新检出的3个错义突变所影响的氨基酸进行了进化保守性分析,4个病例均与GJB2基因相关,但却存在着不同的情况。
1 对象与方法
1.1 对象 本研究纳入2020-05至2022-01于我院产前诊断中心遗传咨询门诊就诊的4例先天性耳聋家系。由患者家属配合完成临床信息采集,包括患者基本信息、耳聋病史、出生史、家族史、个人相关病史(外伤、耳毒性药物使用史、感染史)等。全部纳入参与者均签署书面知情同意书。
1.2 方法
1.2.1 试剂与仪器 TIANamp Genomic DNA Kit离心柱式血液gDNA提取试剂盒(天根,北京),十五项遗传性耳聋基因检测试剂盒(博奥晶芯,成都),xGenExome Research Panel外显子捕获试剂盒(IDT,美国);NovaSeq 6000二代测序平台(Illumina,美国),晶芯EasyArray3A集成化芯片工作站(博奥晶芯,成都),晶芯LuxScanDx24微阵列芯片扫描仪(博奥晶芯,成都),ABI 3730基因分析仪(Thermo Fisher,美国),Bioanalyzer 2100(Agilent,美国),ND 2000紫外分光光度计(Thermo Fisher,美国)。
1.2.2 样本采集和DNA提取 根据所采集家系背景资料,以先证者为核心,使用Cyrillic软件绘制系谱图。采集每位参与成员3 ml EDTA抗凝血,提取总DNA。取2 μl利用紫外分光光度计检测DNA浓度及纯度。
1.2.3 耳聋基因芯片 采用十五项遗传性耳聋基因突变微阵列诊断芯片对纳入的6名家系成员(包括患者1、患者2及其配偶、患者3及其配偶、患者4)进行初筛。该芯片包含了GJB2基因的4个热点突变,即35delG、176del16、235delC、299delAT;SLC26A4基因的8个热点突变,即IVS7-2A>G、2168A>G、1229C>T、1975G>C、1174A>T、1226G>A、2027T>A和IVS15+5G>A;线粒体DNA(mtDNA)的2个热点突变,即1555G>A、1494C>T;以及GJB3基因的538C>T突变。实验过程中的PCR扩增、磁珠捕获分离、磁珠杂交和结果检测的各部分操作均依据厂家提供的标准操作流程进行。
1.2.4 全外显子组测序 对经过1.2.2初筛实验仍无法明确诊断的病例,进行全外显子组测序分析。用xGenExome Research Panel进行外显子区序列杂交捕获,定量荧光PCR的方法测试文库富集情况、大小分布及浓度。NovaSeq 6000平台完成测序;Picard v软件(1.57)去除PCR重复,软件(Burrows-Wheeler Aligner)将原始数据比对人类基因组参考序列(GRCh38版本)。Verita TrekkerVariants Detection systemv2.0(贝瑞基因,中国)和Genome Analysis Toolkit软件进行变异calling;再利用ANNOVAR(v 2.0)和EnlivenVariants Annotation Interpretation系统(贝瑞基因,中国)根据美国医学遗传学与基因组学协会(American College of Medical Genetics and Genomics,ACMG)发布的通用指南进行变异注释解读。
1.2.5 Sanger测序验证 针对各疑似致病的位点利用Primer 5在线软件(网址:http://frodo.wi.mit.edu/primer3)进行引物设计。PCR方法扩增,利用Qiagen纯化试剂盒对产物进行纯化;进行上机测序,系统软件进行结果分析。
1.2.6 错义变异分析 依据先前方法,使用MEGA7在线软件对新检测出的错义变异所影响的氨基酸在各物种间的进化保守性进行分析,全部参数为默认值。同时,利用Revel在线软件对新错义变异的危害性加以评分。
2 结 果
2.1 临床检查 患者1,女,5岁,诊断感音神经性耳聋(双耳)、白甲病、掌跖角化病;其他各项体征均正常,常规实验室血液检测(血常规、血生化等)未见异常。其父母表型正常,否认家族内有耳聋等遗传性疾病患病史,否认患儿致聋性外伤、耳毒性药物使用史、感染史。本例家系图如图1A所示。患者2,女,35岁,出生即诊断先天感音神经性耳聋,其他体征无异常;其妹妹同患先天性耳聋;其丈夫,34岁,出生时听力正常,2岁左右开始听力逐渐下降,直至5岁左右耳聋。患者3,男,2岁,诊断为先天性耳聋、全面发育落后,常规实验室血液检测未见异常。其父母表型均正常。其母亲就诊时孕第二胎7周,要求明确患儿遗传学诊断,并对第二胎进行产前诊断。本例的家系图如图1E所示。患者4,男,33岁,听力下降十余年,因备孕来我中心咨询,要求行遗传学诊断。
2.2 遗传学检测结果 由于耳聋基因芯片的筛查效率有限,不能满足充分明确遗传诊断目的,故利用WES检测,结果均以Sanger测序进行验证。结果显示,4个先天性耳聋病例均与GJB2基因突变相关,然而其具体情况却存在各异性。4个病例的家系图及遗传检测结果见图1;其中,A-B为病例1情况,C-D为病例2,E-F为病例3,G-H为病例4。在病例1中,第II代男性个体为患者(图1A),携带GJB2: c.167T>C杂合突变(图1B),家系验证证明其为新发突变。病例2中,患者2携带MYO7A: c.1214G>A(图1C,左侧)和MYO7A: c.3545A>G(图1C,右侧)的复合杂合突变,这两个突变均为错义突变,且是未报道过的新突变;而患者2的丈夫携带GJB2: c.299delAT(图1D,左侧)和GJB2: c.235delC(图1D,右侧)的复合杂合突变,与患者2为不同基因导致的遗传性耳聋。病例3中,患者3(图1E)存在GJB2: c.257C>G(p.Thr86Arg)(图1F,左侧)和GJB: c.235delC(图1F,右侧)的复合杂合突变;进一步的羊水产前诊断验证发现患者3的同胞胎儿携带c.257C>G突变,而在c.235delC为野生型,仅为杂合子携带者,不会发病。在病例4中,患者4存在GJB: c.109G>A (p.Val37Ile)(图1G)和GJB: c.235delC(图1H)的复合杂合突变,这是导致其后天听力下降的原因。
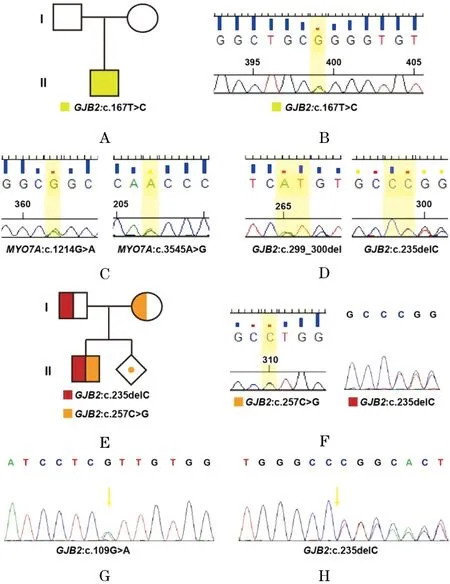
遗传学检测结果细节见表1。如表所示,本研究检出3个与遗传性耳聋相关的新突变体,分别为GJB2: c.167T>C,MYO7A: c.1214G>A和MYO7A: c.3545A>G,扩展了该疾病的突变谱。
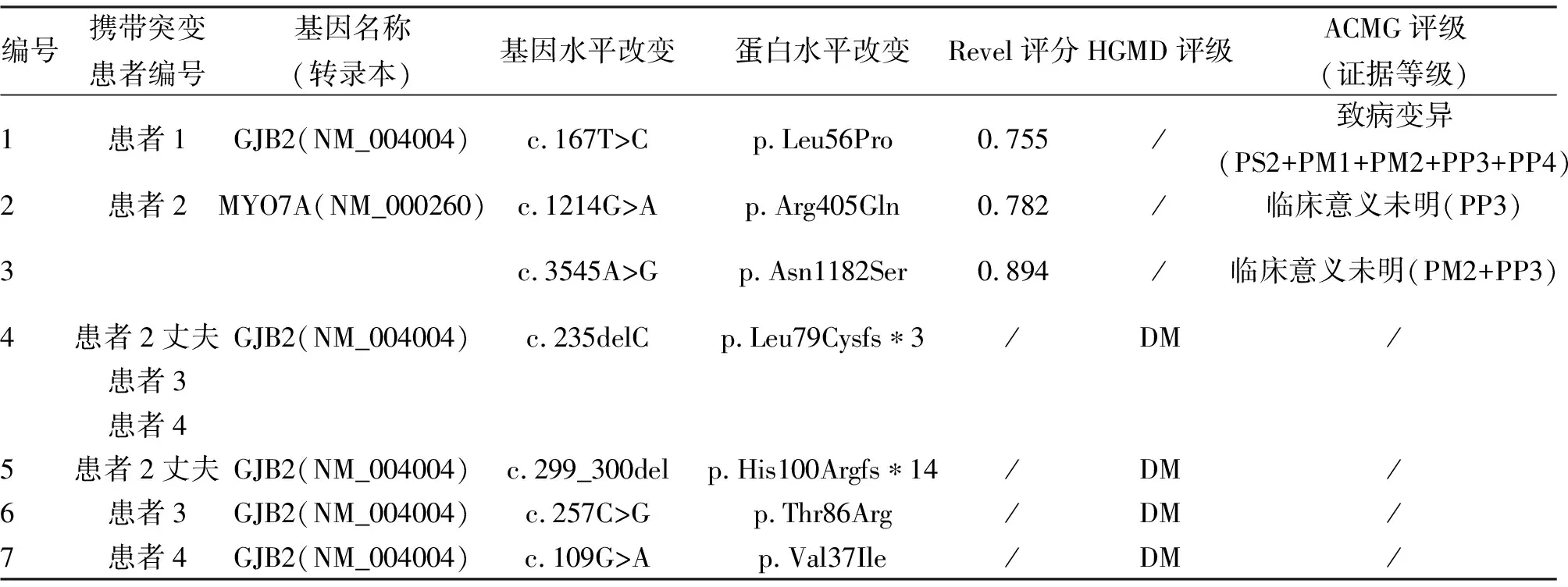
2.3 错义变异分析结果 本研究检出3个新的错义变异,分别是GJB: c.167T>C(p.Leu56Pro)、MYO7A: c.1214G>A(p.Arg405Gln)及MYO7A: c.3545A>G(p.Asn1182Ser)。经MEGA软件分析,这3个变异体所影响的氨基酸在各物种间保持着高度的进化保守性(图2)。Revel评分预测这3个新的错义变异均对蛋白有害(表1)。

3 讨 论
遗传性耳聋在新生儿中的发病率约为1/1000,该类疾病具有很强的遗传异质性,可涉及上百种基因;其中,非综合征性遗传性耳聋约占70%,另外30%为综合征性耳聋,GJB2的基因突变占据非综合征感音神经性耳聋常染色体隐性致病原因的近50%;同时,GJB2突变还可以常染色体显性形式致病,或造成合并有耳聋表型的综合征性疾病。最初,Kelsell等报道了GJB2基因可导致遗传性耳聋。大量的研究揭示了在不同的种族中,该基因存在不同的热点突变分布情况,东亚人群中235delC最常见,欧洲和中东地区35delG最多,W24X在印度人群中最多。从基因型-表型关联角度讲,一般的,截短性突变(无义、移码突变等)会导致更严重的表型,而氨基酸替代(错义突变等)症状较轻。由于GJB2基因有大量频率较高的突变存在,各个突变相互组合后可能产生的预后情况又不尽相同,因此临床中需格外注意。
在本研究中的4例入组家系,经临床评估和遗传检测分析后,证明其均存在GJB2的突变,但均有差异。家系1的患儿临床除了耳聋表型还有白甲病、掌跖角化症,遗传检测发现新发错义突变GJB: c.167T>C(p.Leu56Pro),符合常染色体显性模式。综合判断,该患儿特征较符合Bart-Pumphrey综合征(MIM #148350)。另外,已经有过在此突变临近位置的错义突变导致Bart-Pumphrey综合征的报道,佐证了上述判断。一般情况下,该患儿的父母再次生育受累个体的概率较低,但不能完全排除父母存在生殖腺细胞嵌合的情况。家系2中,夫妻双方虽然均为先天聋人,但两人属于在不同的基因上存在复合杂合突变,孕育先天耳聋患儿的机会非常低,不建议依据双方所携带的突变去进行产前诊断,而是采取常规产检即可。患者2携带MYO7A基因的两个新突变,且其致病性受到生物信息学分析结果的支持,这个发现扩充了该基因的突变谱。家系3为典型的GJB2复合杂合突变导致常染色体隐性先天性耳聋的情况,对胎儿羊水细胞进行GJB2基因测序验证,结果显示其带有c.257C>G杂合突变,应当为无症状携带者,建议继续妊娠。家系4属于GJB2复合杂合突变导致常染色体隐性迟发性耳聋,这在一定程度上反映了GJB2: c.109G>A突变在临床咨询中的挑战。由于该突变存在人群频率高、表型异质性强的问题,常与迟发性耳聋相关。因此,在临床咨询中,应当就此为患者进行详细解释,对再次妊娠说明情况,尊重夫妻双方的选择。
本研究对4例先天性耳聋相关病例进行了详细的遗传诊断及咨询,明确了其不同的发病原因。同时,检出了一个GJB2基因新突变,两个MYO7A基因新突变,扩充了遗传性耳聋的突变谱,为后续研究和临床诊疗实践奠定了基础。本研究样本量较小,无法充分反映GJB2基因相关耳聋的临床表型多样性之全貌,专科医院在监控先天性耳聋病例的诊疗全过程无法覆盖患者初诊、产前筛查及诊断、治疗及妊娠处置的全过程,会造成部分信息丢失。笔者将继续关注该疾病,进一步开展研究工作。
