CD38 deficiency activates ERK1/2-NF-κB signaling pathway in sepsis-associated renal injury
2022-06-30ZHANGHuiqingDUYunaXIEZhengyuWANGZeyuLIHuaLIRong
ZHANG Huiqing,DU Yuna,XIE Zhengyu,WANG Zeyu,LI Hua,LI Rong
(1.Department of Clinical Laboratory, Jiangxi Provincial People's Hospital & The first Affiliated Hospital of Nanchang Medical College, Nanchang, China; 2.Department of Clinical Laboratory, the Second Affiliated Hospital of Nanchang University & Jiangxi Provincial Key Laboratory for Laboratory Medicine, Nanchang, China)
Abstract: CD38 is known to play roles in various inflammatory pathways. However, whether it has a protective or detrimental effect during bacterial septicemia remains disputed. Herein, this study aimed to determine the potential effect of CD38 on renal injury in septicemia. Escherichia coli (E. coli) was used to induce sepsis-associated renal injury in mice. WT and CD38-/- mice were stimulated with E. coli. After three hours, the serum was collected to detect renal function. Function mRNA expressions inflammatory cytokines in kidneys were quantified by real-time PCR. Hematoxylin and eosin staining were used to observe the histomorphology of kidney. The expression of TLR4, NF-κB, MAPK and cytokines were detected by Western Blot. Our results demonstrated that 3×108 cfu/mL E. coli is the appropriate dose to induce sepsis mice model. Compared to WT sepsis mice, CD38-/- mice showed aggravated kidney injuries with impaired renal function, increased inflammation and apoptosis after E. coli stimulation. Interestingly, CD38 deficiency also led to elevated expression of TLR4 and increased phosphorylation of NF-κB p65/p105 and ERK1/2. To sum up, our results suggested that CD38 deficiency could aggravate E. coli-induced renal injury through activating ERK1/2-NFκB signaling pathway.
Key words: CD38; ERK1/2; NF-κB; sepsis; renal injury
1 Introduction
Bacterial infections play an important role in clinical inflammation -related diseases, especially sepsis. After bacterial infection, a series of inflammatory reactions happened, including increased leukocyte count, quickened pulse, and accelerated respiratory rate[1], these reactions would lead to multiple organ dysfunction syndrome(MODS) and sepsis[2-3]in severe cases. Most of the symptoms of infection are caused by Gram-negative (G-) bacteria, such as Escherichia coli(E.coli)[4]. Lipopolysaccharide (LPS)of G-Bacteria could induce inflammatory response by activating toll-like receptor 4 (TLR4), which is involved in the classic inflammatory pathway[5].
Cluster of differentiation 38 (CD38), also known as cyclic ADP ribose hydrolase, is a glycoprotein that exists on the surface of many immune cells such as T cells, B lymphocytes, dendritic cells (DCs),macrophage and natural killer cells[6]. It is involved in the activation[7], proliferation[8], and survival[9]of human and mouse lymphocytes. In immunocytes,CD38 is involved in regulation of different biological reactions, such as protein phosphorylation[10], cytokine secretion[11]and apoptosis[12]. Studies have shown that CD38 is abnormally expressed during in fection, and its absence can alter the migration patterns of neutrophils and monocytes[6,13]in inflammatory tissues and increased susceptibility to infection[14].
The toll-like receptor (TLR) family is composed of highly conserved transmembrane receptors that are part of the innate immune system[15-16]. They are expressed by immune[17]and non-immune cells, such as cardiomyocytes[18]. TLRs participate in the recognition of pathogen -associated molecular patterns(PAMPs)[19]and the activation of inflammatory pathways which regulate gene expression, thus stimulating the production of inflammatory factors such as tumour necrosis factor-α (TNF-α), interleukin-1β(IL-1β), and inducible nitric oxide synthase(iNOS)[20].After LPS recognition, TLR4 can activate nuclear factor-κB(NF-κB) and other downstream intracellular signals. Pathogenic microorganisms activate the NF-κB signalling pathway through TLR4 in immune cells such as macrophages, dendritic cells, and B cells[21]. Numerous researches have suggested that,TLR4/NF-κB is one of the most important inflammatory pathway in vivo[10,22-23].
Mitogen-activated protein kinase (MAPK) is a group of serine-threonine protein kinases that can be activated by different extracellular stimuli, such as cytokines[24], neurotransmitters, growth factors, hormones, cell stress[25]and cell adhesion[26]. MAPK is widely expressed in eukaryotic cells, and is the basic component of conservative 3 levels kinases cascades from yeast to human, including the MAP kinase kinase kinase(MKKK), MAP kinase kinase(MKK) and MAPK. These three kinases can be activated successively, and induce the regulation of cell growth,cell differentiation and cell stress adaption to the environment[27], inflammation[28]and apoptosis[29].MAPK has three main components: extracellular regulated protein kinases1/2(ERK1/2), c-Jun N-terminal kinase (JNK) and p38. During ischemic and septic renal injury, ERK1/2 is activated and phosphorylated rapidly[30]. And previous studies also have shown that, LPS stimuli-induced inflammatory responses activated NF-κB signalling via the phosphorylation of ERK1/2[31].
Our previous studies showed that TLR4 expression significantly increased in CD38-/-mice compared with wild-type (WT) mice. However, whether CD38 has effects on MAPK in sepsis-associated renal injury are still unknown. Hence, this study is to explore the role of CD38 in the sepsis-associated renal injury triggered by bacterial infection.
2 Materials and Methods
2.1 Animals
Wild type mice (C57BL/6) were obtained from the Laboratory Animal Center of Wuhan University,and CD38 deficiency (CD38-/-) mice (B6.129P2-Ly96-/-) were donated by Professor Hongbo Xin and Professor Keyu Deng of Translational Medicine Institute of Nanchang University. Male eight-weeks mice (20±2 g) were selected for all experiments. All experiments were approved by the Animal Protection Committee of Nanchang University.
2.2 Preparation of bacterial and establishment of mice model
To establish bacterial infection sepsis model,standard E. coli strain ATCC25922 (G-bacteria)were used, which was kindly offered by Jiangxi King -Med Microbiology Laboratory. In brief, the bacterial was seeded into Columbia blood medium and then cultured in a 37 ℃incubator for 12 hours.According to a report by Sun et al[32], the intraperitoneal injections of 0.5 mL E. coli in dose with 3×108cfu/mL could induce sepsis in mice. Based on this, to evaluate the appropriate injection concentration, we infected mice with 0.5 mL E. coli (1×108cfu/mL or 3×108cfu/mL) and 0.5 mL PBS as control by intraperitoneal injection. Three hours after injection, the blood and kidney tissues were collected for further experiments.
2.3 Serum analysis
Blood was obtained and centrifugated to collect serum. Next, the serum creatinine(Scr) concentration, blood urea nitrogen(BUN), and uric acid(UA) were detected with Creatinine reagent kit (Biote, Nanchang, China), Urea reagent kit (Biote, Nanchang,China), and Uric Acid reagent kit (Meikang biotech,Ningbo, China) respectively, measured by Automatic measure biochemical analyzer AU5800 (Beckman Coulter, USA) in the Clinical Laboratory of the Second Affiliated Hospital of Nanchang University.
2.4 Bacterial culture of kidney tissues and gram staining
A total of 100 mg of mouse kidney were taken and grind it into homogenate. After that, 100 μL homogenate was taken out and painted on agarose medium evenly, and then cultured in 37℃for 24 hours with an incubator. Then, pick out a single colony for Gram staining and observe it under the microscope.
2.5 Histological staining
The kidneys were fixed in 4% paraformaldehyde (PFA) overnight, and washed with PBS (pH 7.4)3 times. And after that, the tissues were dehydrated in a gradient ethanol (60%, 75%, 85%, 95% twice,and 100% twice), and rendered transparent with xylene and embedded. Paraffin sections of 3 μm were prepared using a microtome. The sections were heated at 65 ℃for least 2 hours for deparaffination and hydrated by submersion in the following solutions:xylene for 5 minutes three times, 100% ethanol for 30 seconds twice, 95% ethanol and 70% ethanol for 30 seconds. Then, sections were stained with hematoxylin and eosin (H&E) and mounted on glass slides.
2.6 RNA extraction and real-time quantitative PCR assay
The total RNA from kidney was extracted by using TRizol reagent (Invitrogen, US) and then treated with gDNA Eraser (Takara Biotech, Japan) at 42℃ for 2 minutes. RT-PCR was performed using PrimeScriptTMRT under the following conditions: 37℃for 15 minutes, 85 ℃for 5 seconds, and 4 ℃indefinitely for product preservation.
The next day, beside the road, the red flowers were brighter than the day before. The natural beauty made me gasp4. It gave me an understanding of the energy of growing and youth and convinced me it couldn t be destroyed with a heartless stick. I wanted to be like the soul of a flower.
RT-qPCR was performed using SYBRPremix Ex TaqTM Ⅱ (Tli RNaseH Plus) and the StepOneTMPLUS Real-Time PCR System (Applied Biosystem,New York, USA). And the sequence of primers we used were shown in Table 1.We detected the threshold cycle (Ct) for all genes and determined their relative expression levels compared to GAPDH.
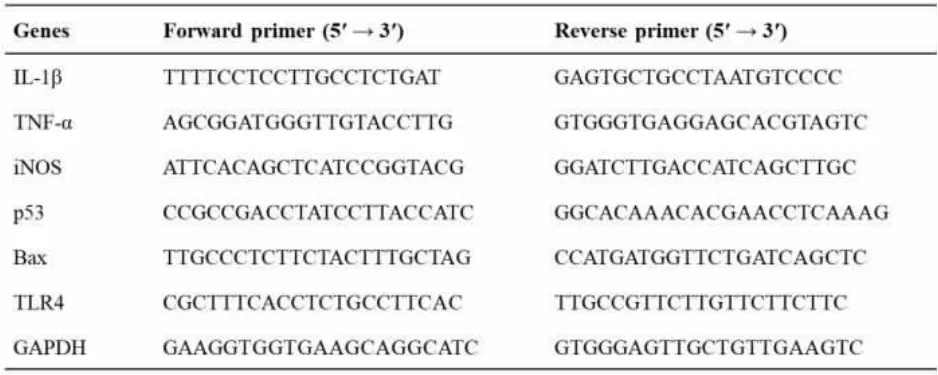
Table 1 The sequence of primers for RT-qPCR
2.7 Western blot assay
The protein expression of TLR4, IL-1β, TNFα, iNOS, p53, Bax, MAPK and NF-κB in kidney were detected by Western blot. In brief, the kidney tissues protein was extracted by RIPA Lysis Buffer with Phenylmethanesulfonyl fluoride (PMSF) and loaded into 10% SDS-PAGE gel and transferred onto PolyVinylideneFluoride (PVDF) membranes. The primary antibodies of anti-TLR4 (1:500, CST, USA),anti-IL-1β (1:500, Boster, China), anti-TNF-α (1:500, Boster, China), anti-iNOS(1:1 000, CST, USA),anti-p53 (1:2 000, Proteintech, USA), anti-Bax (1:5000, Proteintech, USA), anti-p38 (1:1 000, CST,USA), anti-Phospho-p38 (1:1000, CST, USA), anti-ERK1/2 (1:1 000, CST, USA), anti-Phospho-ERK1/2 (1:2 000, CST, USA), anti-JNK (1:1000, CST, USA), anti-Phospho-JNK (1:1 000, CST, USA), anti-NF-κB p65 (1:1 000, CST, USA), anti-Phospho-NF-κB p65 (1:500, CST, USA), anti-NF-κB p105(1:1 000, CST, USA), anti-Phospho-NF-κB p105 1:500, CST, USA) and anti-GAPDH (1:5000, Proteintech, USA) were used. The secondary antibody incubation dilution ratio was goat anti-rabbit (1:1 000,Zhongshan Biotech, Beijing, China) and goat antimouse (1:1 000, Zhongshan Biotech, Beijing, China).The protein expressions were visualized by the ECL assay (Sage creation) according to the manufacturer’s instructions.
2.8 Statistical analysis
All data in this study were expressed as mean ±standard deviation (SD). Paired t-test was used to determine the statistically significant difference between the two groups, and one-way ANOVA and Tukey post-test were used to estimate the difference between multiple groups. And if P<0.05, the difference is considered significant. All analyses were performed using the software GraphPad Pro 5.0(GraphPad, San Diego, CA, USA).
3 Results
3.1 3×108 cfu/mLE. coli induced WT mice showed severe illness and abnormal renal function
It has been reported that, three hours after 3×108cfu/mL E. coli injection can induce sepsis-associated organ injury[33]. To further confirm the minimal amount required to induce sepsis-associated renal injury, we injected 1×108cfu/mL E. coli, 3×108cfu/mL E. coli, or PBS into WT mice intraperitoneally.After 3 hours of infection, PBS-injected WT mice(Figure 1a) showed no apparent behavioral changes while WT mice injected with 1×108cfu/mL and 3×108cfu/mL E.coli showed symptoms of sickness such as rough fur and fatigue (Figure 1b and 1c). The symptoms were more prominent in WT mice injected with 3×108cfu/mL E. coli. Also, WT mice injected with 3×108cfu/mL E. coli had significant higher concentrations of Scr (Figure 1d), BUN (Figure 1e),and UA (Figure 1f) than PBS-injected WT mice or with 1×108cfu/mL E.coli-injected mice. Mice injected with 1×108cfu/mL E. coli showed significantly increased BUN (Figure.1e), but no significant elevation of Scr (Figure.1d) or UA (Figure 1f) when compared with PBS-injected WT mice. Taken together,these data suggested that 3×108cfu/mL E. coli is necessary to induce severe illness and abnormal renal function in WT mice.
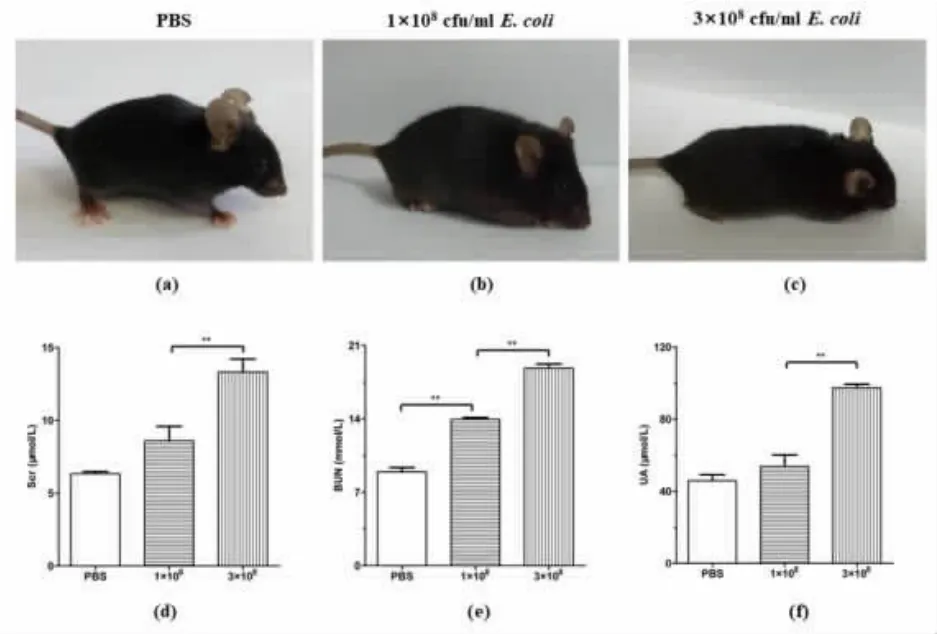
Figure 1 3×108 cfu/mL E. coli induces severe illness and abnormal renal function in WT mice
3.2 E. coli induces expressions of TLR4, pro-inflammatory cytokines and pro -apoptotic genes in kidney tissues of WT mice
The inflammatory reactions of kidney tissues in 3×108cfu/mL E. coli-stimulated mice were more severe than those of PBS-stimulated WT mice(Figure.2a, 2b, 2c and 2d). Mice induced with 3×108cfu/mL E. coli presented distinct renal injuries, including tubular lumen narrowing and glomerular congestion(yellow arrows), interstitial edema(black arrows), and a severe infiltration of inflammatory cells in the glomerulus of kidneys(blue arrows), while PBS-stimulated WT mice showed no injuries (Figure 2a, 2b,2c and 2d).
To confirm that the kidney injuries were induced by the injected E. coli, we cultured kidney tissues from PBS- and 3×108cfu/mL E. coli-stimulated mice and identified the cultured bacteria with Gram staining. Our data showed no bacterial growth in the kidneys of PBS-stimulated mice (Figure 2e),while one type bacterial colony appeared in the E.coli-stimulated group (Figure 2f). Furthermore, the Gram staining indicated that the colony was formed by simplex G-bacteria(Figure 2g).
To further explain the aggravated inflammation in E. coli-stimulated WT mice, we measured the mRNA levels of TLR4 (Figure 2h) and the following pro-inflammatory cytokines in kidneys: TNF-α (Figure 2i), iNOS(Figure 2j) and IL-1β(Figure 2k). Also the mRNA levels of pro-apoptotic genes: p53(Figure 2l) and Bax (Figure 2m) were measured. Our data showed significantly higher expressions of all the cytokines, pro-apoptotic factors and TLR4 in the kidneys of E. coli-stimulated WT mice than in the PBS-stimulated group, suggesting a stronger inflammatory response occurred in E. coli-stimulated WT mice kidneys, and also increased expression levels of apoptotic genes.
3.3 CD38-/- mice present aggravated renal injuries with E. coli stimulation
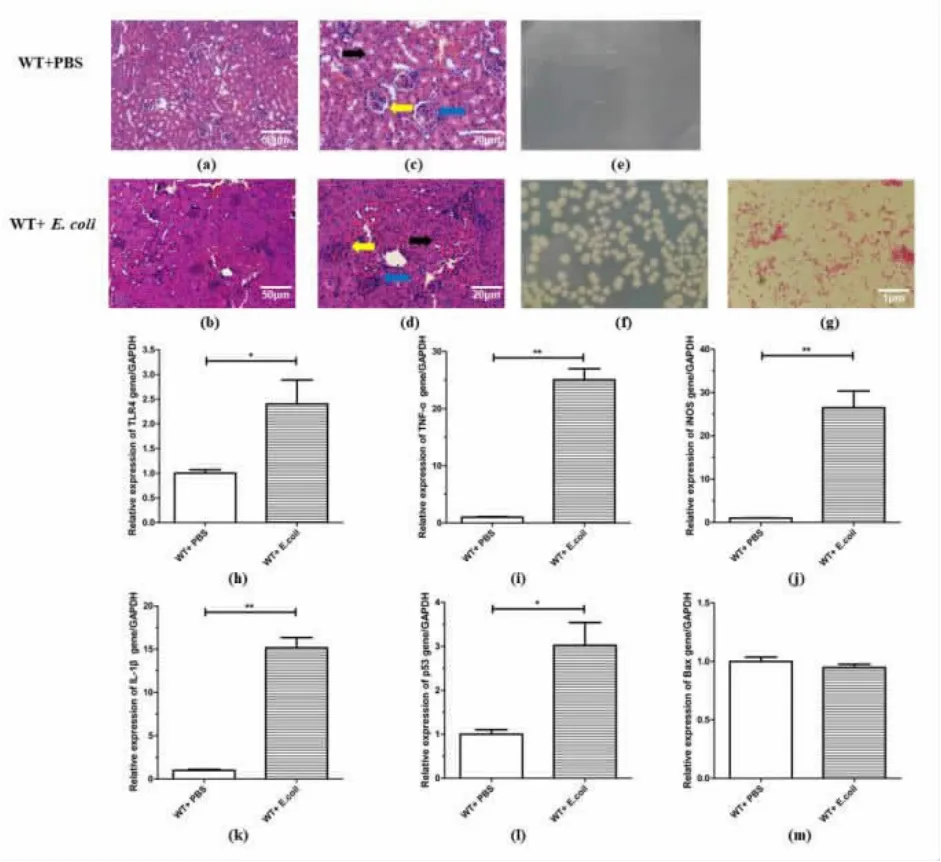
Figure 2 Effects of E. coli on gene expressions of TLR4,pro-inflammatory cytokines and pro-apoptosis genes in kidneys of WT mice

Figure 3 Effect of CD38 deficiency on E. coli induced renal injuries in mice
3.4 E. coli stimulation enhanced expressions of cytokines and pro-apoptotic proteins in kidneys of CD38 deficient mice
To study the effects of CD38 deficiency on proinflammatory cytokines production, the mRNA expressions of IL-1β(Figure 4a), iNOS(Figure 4b) and TNF-α (Figure 4c) were measured in kidneys of WT and CD38-/-mice at 3 hours after E. coli stimulation.IL-1β and iNOS increased significantly in CD38-/-mice when compared to WT mice, but TNF-α had no significant change. Moreover, Western Blot results showed that CD38 deficiency significantly increased IL-1β(Figure 4f and 4g) and iNOS(Figure 4h and 4i)expressions. And, there was no difference in TNF-α protein expression between these two groups either(Figure 4j and 4k). Furthermore, pro-apoptotic proteins were detected to observe renal apoptosis in WT and CD38-/-mice induced by E. coli. In CD38-/-mice, the mRNA expressions of p53 (Figure 4d) and Bax (Figure 4e) were significantly higher than in WT mice. And the protein levels of p53 (Figure.4l and 4m) and Bax (Figure.4n and 4o) showed the same trend. These results suggested that, the deficiency of CD38 can cause the elevation of pro-inflammatory factors and aggravate inflammatory response. And at the same time, up-regulating the expressions of proapoptotic factors lead to more apoptosis of kidney cells. Anabatic inflammatory and apoptotic reactions resulted in more severe renal injuries in E. coli-induced CD38-/-mice.
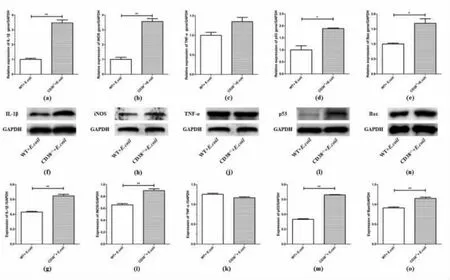
Figure 4 Expressions of pro-inflammatory cytokines and pro-apoptotic protein in kidneys of E.coli-induced WT and CD38-/- mice
3.5 CD38 deficiency aggravated renal injury through activating ERK1/2 signaling pathway
To further reveal the mechanism of aggravated renal injuries in CD38-/-sepsis mice, we investigated the MAPK signaling pathway, which plays an important role in inflammatory response. Western blot was performed to detect expression levels of the three main molecules (p38, JNK, ERK1/2) of MAPK pathway. The results manifested that, in kidneys of WT and CD38-/-mice, the protein levels of p38(Figure 5a and 5b) and JNK(Figure 5c and 5d) and their phosphorylation did not change significantly compared to WT mice. And what makes sense is that compared with WT mice, CD38 deficient leads to significantly higher expressions of phosphorylated ERK1/2, while there was no difference of ERK1/2 levels between two groups(Figure 5e and 5f). These results suggested that CD38 deficiency aggravated renal injury through activating ERK1/2 pathway.

Figure 5 The protein expressions of MAPK signaling pathway in kidneys stimulated by E. coli
3.6 CD38 deficiency aggravated renal injury through NF-κB signaling pathway
The NF-κB signaling pathway also plays an important role in the inflammatory response when TLR4 is activated. To further explore the function of NF-κB signaling pathway of sepsis-associated renal injuries, we detected mRNA and protein levels in kidneys of two stimulated groups. In kidneys of E.coli stimulated mice, CD38 deficiency could significantly increase the mRNA expression of TLR4(Figure 6a), but there was no significant difference in protein level of TLR4(Figure 6b and 6c). The protein levels of NF-κB p65 showed no difference(Figure 6d), but the expression of phosphorylated NF-κB p65 (Figure 6d) were significantly increased in CD38-deficient mice and the ratio of Phospho-NFκB p65/p65 (Figure 6f) was significantly higher than the other group. And the expressions of NF-κB p105, phosphorylated NF-κB p105 showed the same trend(Figure 6e and 6g). These results indicated that,the deletion of CD38 could activate NF-κB signaling pathway and aggravate renal injuries caused by bacterial infection.
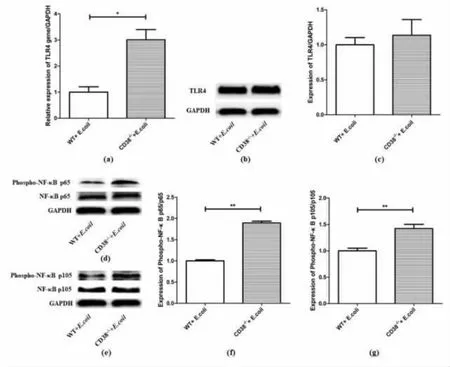
Figure6 Expression levels of TLR4 and NF-κB p65/p105 in kidneys of E. coli-induced WT and CD38-/- mice
4 Discussion
CD38 is an essential molecule that is involved in lymphocyte activation and the defense against infectious diseases[34]. CD38 plays different roles in different organs and diseases, whether CD38 deficiency can protect or aggravate the organ damage caused by sepsis remains controversial. Previous studies indicated that, the blockade of CD38 leads to reduced M1 macrophage activation, thus, protecting kidney and spleen from acute injury by diminishing the NF-κB signaling pathway[35]. Additionally, inhibition of the CD38/cADPR pathway protected rats from septic brain damage[36], while inhibition of the NAD+/CD38/cADPR/Ca2+signaling pathway protected heart, liver, and kidney from relevant injury[37]. However, some researches showed that CD38 deficiency resulted in increased susceptibility to several pathogens, such as Listeria monocytogenes (L. monocytogenes)[6], Mycobacterium avium (M. avium)[38]and Streptococcus pneumoniae (S. pneumoniae)[39], and the parasite Entamoeba histolytica (E. histolytica)[13].Also CD38 contributes to pro-inflammatory phenotypes in innate immune cells such as macrophages and dendritic cells[14].
Analogously, in this study, we demonstrated that CD38 deficiency significantly aggravated the inflammatory reaction and elevated pro-inflammatory cytokines expressions in an E. coli-induced murine model of sepsis. In concordance with our findings,other researchers have reported that CD38+CD4+T cells are effective in reducing blood parasite burden during the human innate immune response against malaria[40], and an increase in CD19+CD38hiCD24hitransitional B cells protects against neonatal sepsis[41].Also, it is reported[42]that CD38 inhibits cell apoptosis and promotes proliferation in cervical cancer cells. In our research we found that in bacteria-induced septicemia mice, CD38 deficiency could aggravate renal injuries by up-regulating expressions of pro-apoptotic molecules. As a response to infection, the role played by CD38 varies. Therefore, its specific functions and related modulators are worth exploring in different scenarios.
Our study focused on bacteria-induced renal injuries in a septic mice model. E. coli, a common G- bacterium, causes sepsis through LPS. Therefore,researchers have paid special attention to LPS-induced sepsis and discovered certain protective molecules. For example, corylin, isolated from Fabaceae plants, showed anti-inflammatory properties on LPS-induced sepsis and inflammatory reactions[43], and harmine also could mitigate LPS-induced acute kidney injury[44]. A clinical research reported that, levels of IL-1β were shown to be higher in patients who died than in those who survived during sepsis, suggesting an association between high levels of IL-1β and sepsis outcome[45]. This is consistent with our findings that IL-1β expression was higher and inflammatory response was more severe in CD38-/-mice.
Studies have shown that NF-κB activation can induce transcription of many genes, associated with not only inflammatory but also apoptotic responses[31].In this study, phosphorylated NF-κB p65/p105 levels in kidneys were significantly increased in CD38-/-mice compared to WT mice at 3 h after E. coli injection, and our RT-qPCR and western blot studies revealed high expressions of pro-apoptotic molecules including p53 and Bax. Research suggests that the p53 -CypD complex mediates renal tubular cell apoptosis in ischemic renal injury[46]and c-Myc triggers a proapoptotic mitochondrial destabilizing activity cooperating with Bax[47]. In line with these findings, our results showed that the expressions of p53 and Bax were higher in CD38-/-mice than WT mice with E. coli injection respectively. Moreover, inhibition of apoptosis can significantly increase survive and improve organ damage caused by sepsis[48]which is consistent with our study that higher expressions of pro-apoptotic proteins can lead to more severe renal injury, elevated renal function indicators (Scr,BUN and UA), renal dysfunction and even renal failure.
And other studies have proved that blocking MAPK signaling pathway can significantly improve the renal injury caused by sepsis[49], indicating that MAPK signal transduction is involved in the occurrence and development of sepsis. And ERK1/2 was identified as a regulator of kidney injury molecule-1(KIM-1) expression[30]. Consistent with the above study, our results showed that CD38 knockout can up-regulate the phosphorylation of ERK1/2, aggravating the renal injury of mice. Research has confirmed that DC-specific ICAM-3-grabbing nonintegrin (DC-SIGN) led to the upregulation of inflammatory cytokines via activating ERK1/2-NF-κB/p65 signaling pathway[31]. And specific inhibition of ERK1/2 reduced the nuclear accumulation of NF-κB p65, phosphorylation of NF-κB p65 and reversed the elevated phosphorylation of cytosolic NF-κB p65[50]. These studies indicate that ERK1/2-NF-κB signaling pathway plays an important role in the regulation of inflammation. In this study, our results showed that the phosphorylation level of ERK1/2 in CD38 deficient mice stimulated by E. coli was significantly higher than that in WT mice, and the expression of NF-κB p65/p105 in the nucleus was increased and significantly increased the phosphorylation of NF-κB p65/p105. Activation of ERK1/2-NFκB signaling pathway caused by CD38 deficiency resulted in more serious renal injuries.
Collectively, in our research, we proved that CD38 plays an important role in bacterial septicemia. The absence of CD38 aggravated renal injury in septic mice, which leads to violent inflammation and apoptosis reactions by activating ERK1/2-NF-κB signaling pathway. And also, CD38 deficiency led to increasing the susceptibility of organism to bacteria,resulting in more serious inflammatory response and injury.
Conclusions
According to the experimental results of this study, we can conclude that CD38 plays an important role in septic mice, and CD38 deficiency mainly activates ERK1/2-NF-κB signaling pathway, which aggravates the renal injury of septic mice. Therefore,ERK1/2 inhibitors or CD38 activators may be used as potential drugs to reduce E. coli induced sepsis related renal injury.
Data Availability
The data used to support the findings of this study are available from the corresponding author upon request.
Conflicts of Interest
The authors declare that there are no conflicts of interest.
Author’s Contributions
#These two authors, Huiqing Zhang, and Yuna Du contributed equally to this work. ZX, ZW and HL carried out the experiments, and RL participated in the project design, coordination the experiments, and helped to draft the manuscript. All authors read and approved the final manuscript.
Acknowledgments
This work was supported by the National Natural Sciences Foundation of China (NSFC) grant numbers [31960165&81760288].
