基于生物信息学分析CCNB1、ASPM 和AURKA 作为肝细胞癌预后标志物的潜力
2022-06-30蒲俊兴何颖陈娟娟
蒲俊兴,何颖,陈娟娟
(1.南昌大学第二附属医院检验科 江西省检验医学重点实验室,江西 南昌 330006;2.南昌大学公共卫生学院,江西 南昌 330031)
肝细胞癌(HCC)是世界癌症相关死亡的第二 大常见原因[1],由于早期诊断困难,大部分患者在入院检查时已进入中晚期[2]。 目前的治疗手段包括手术切除、肝移植、介入治疗和分子靶向,而首选治疗方式是手术治疗。 尽管肝移植和手术切除等治疗可用于早期肿瘤,但只适用于少数患者。 对于晚期患者,仅适用于支持性治疗,而其中大多数患者对常规化疗或放疗耐药[3]。HCC 的发病率正在上升,预计在不久的将来还会继续增长[4]。 因此,探索HCC 的预后标志物是有必要的。
近年来, 生物信息学和多组学分析广泛应用于肿瘤发生和发展过程中基因表达变化的研究。随着大数据时代的到来, 通过对各种生物医学公共数据库进行数据挖掘,可以方便、快捷地发现具有差异表达的基因(DEGs),从而找到疾病治疗相关的潜在靶标以及预后评估的可能标志物。 本研究从GEO 数据库下载了HCC 患者的数据, 通过DEGs 构建了PPI 网络。 筛选出枢纽基因并验证其在HCC 患者中的转录、 翻译和生存情况。 利用TIMER 数据库评估枢纽基因的表达水平及其与肿瘤纯度和浸润免疫细胞的相关性。
1 材料和方法
1.1 数据来源及处理 从GEO 数据库(https://www.NCBI.nlm.nih.gov/GEO/) 下载两个肝细胞癌芯片数据(GSE14520[5]和GSE46408[6])。 GSE14520 基 于GPL3921 平台, 包括225 例HCC 样本和220 例正常肝脏样本;GSE46408 基于GPL4133 平台, 包括6 例HCC 样本和6 例正常肝脏样本。 GEO2R 软件(https://www.ncbi.nlm.nih.gov/geo/geo2r)筛 选HCC 和正常组织之间的DEGs。 校正后P<0.05 和log2FC≥1.0 的基因被认为是DEGs。 利用Venny 2.1.0 工具(https://bioinfogp.cnb.csic.es/tools/venny/index.html)展示DEGs。
1.2 构建PPI 网络及筛选枢纽基因 STRING 数据库(http://string-db.org/)用于构建PPI 网络。 将筛选出的DEGs 输入STRING 中,提取与综合评分>0.4配对的PPI 网络。 通过Cytoscape 软件对其进行可视化,利用CytoHubba 插件计算蛋白质节点的连接度。 连接度较高的基因被视为枢纽基因。
1.3 验证枢纽基因在HCC 中差异表达 ONCOMINE 数据库(http://oncomine.org)为全基因组表达分析提供了重要的生物信息学帮助[7]。 利用其分析HCC 中枢纽基因的表达情况,P<0.05 为差异有统计学意义。
1.4 枢纽基因的预后价值 GEPIA2(http://gepia2.cancer-pku.cn/#index)工具中共529 例肝脏样本数据(包括HCC 样本479 例,正常肝脏样本50 例),对枢纽基因在低表达组和高表达组中的预后价值进行分析。
1.5 枢纽基因的变异分析 cBioPortal(https://cbioportal.org)是一个能够可视化和分析多维癌症基因组数据的工具[8]。 利用其评估HCC 中拷贝数变异(CNV)、 突变和基因类型并分析基因突变与HCC预后的关系。
1.6 枢纽基因共表达网络的构建及功能富集分析GeneMANIA(http://www.genemania.org)是用于分析靶基因的遗传和蛋白质相互作用、共表达、通路、共定位和结构域蛋白相似性的在线数据库[9]。 利用该数据库分析枢纽基因及其互作基因之间的关系。
DAVID 6.8 (https://david.ncifcrf.gov/home.jsp)能够很好地阐明目的基因生物学功能[10]。利用其对共表达网络进行GO 功能和KEGG 通路富集分析。使用R 软件中“ggplot2”包对富集的功能和通路进行展示。
1.7 肿瘤免疫浸润分析 TIMER(https://cistrome.shinyapps.io/timer/) 可用于系统评估各种免疫细胞的浸润及其临床影响[11]。本研究对枢纽基因在HCC中的表达水平及其与肿瘤纯度和浸润性免疫细胞的相关性进行分析。 此外,还评估了基因表达与临床结果和免疫细胞浸润之间的相关性。
2 结果
2.1 DEGs 的鉴定 利用GEO2R 从GSE14520 中共鉴定出202 个DEGs, 其中上调基因104 个,下调基因98 个,见图1a;从GSE46408 中鉴定出156个DEGs,其中上调基因99 个,下调基因57 个,见图1d。 对DEGs 进行Venny 2.1.0 分析,得到DEGs的交集。 交集的DEGs 共有46 个,其中25 个表达上调,21 个表达下调,见图1b—图1e。
2.2 PPI 网络构建与枢纽基因筛选 通过STRING构建DEGs 的PPI 网络涉及45 个节点和280 个边,见图1c。 使用Cytoscape 软件对该PPI 网络进行可视化并利用CytoHubba 插件计算每个蛋白质节点的连接度。 本研究选取连接度排名前三的基因(CCNB1、ASPM 和AURKA)为枢纽基因,见图1f。 这3 个枢纽基因在HCC 中均上调。

图1 GEO 两个数据集中的DEGs
2.3 HCC 患者中枢纽基因的异常表达 在ONCOMINE 和GEPIA2 数据库中获取CCNB1、ASPM和AURKA 在HCC 中的异常表达数据。 两者结果均表明, 与正常样本相比,HCC 中CCNB1、ASPM和AURKA 的表达水平或转录水平显著升高 (P<0.05),见图2a—图2b。
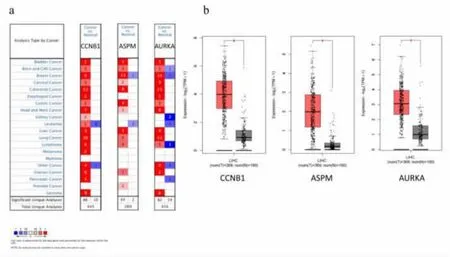
图2 枢纽基因在不同组织中的表达
2.4 HCC 患者中枢纽基因的预后分析 使用GEPIA2 数据库评估这3 个枢纽基因与HCC 预后之间的关系。 数据显示,CCNB1、ASPM 和AURKA的高转录水平与HCC 患者较短的OS 显著相关(P<0.05),见图3a—图3c。 此外,还研究了枢纽基因在HCC 患者DFS 中的预后作用。结果表明CCNB1、ASPM 和AURKA 的高转录水平与HCC患者较短的DFS 显著相关(P<0.05),见图3d—图3f。
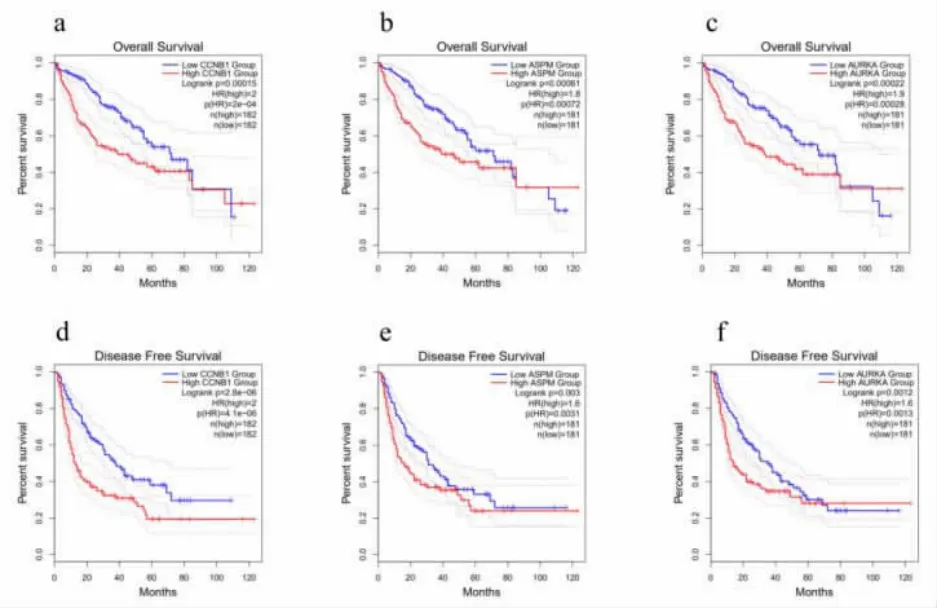
图3 HCC 中枢纽基因的生存分析
2.5 HCC 患者中枢纽基因的改变频率分析 使用cBioPortal 分析HCC 患者中3 个枢纽基因的遗传改变频率。 如图4a 所示,98 名(25.26%)HCC 患者的这3 个基因发生了明显的改变,包括扩增、深度缺失、截短突变、错义突变和转录上调。 具体而言,CCNB1、ASPM 和AURKA 遗传改变的百分比变化分别为8%、16%和9%,见图4b。 我们还比较了它们的表达变化与HCC 预后之间的关系。结果表明,HCC 患者中枢纽基因的改变与OS 显著相关 (P=0.0229),见图4c。 与不改变枢纽基因表达相比,改变枢纽基因表达的HCC 患者表现出更差的DFS(P=0.0256),见图4d。
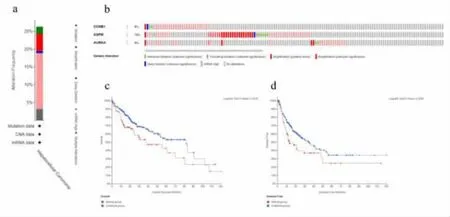
图4 LIHC 中枢纽基因的改变频率及其生存分析
2.6 枢纽基因的共表达和功能富集分析 共表达网 络 显 示CDK1、DLGAP5、FZR1、CPEB1、BORA、TPX2、CCNF、TACC3、PLK1、PTCH1、NDC80、AURKAIP1、WEE1、KIF3A、CDC25A、FOXO1、PPP6C、HMMR、BIRC5、CDC25B 这20 个 基 因 与CCNB1、ASPM 和AURKA 的调控功能相关,见图5a。 利用DAVID 6.8 分析CCNB1、ASPM 和AURKA 及其互作基因的功能。图5b-图5d 结果显示,生物学过程(BP)主要富集在有丝分裂(核分裂)、有丝分裂细胞周期的G2/M 转变、细胞分裂、后期促进复合物依赖的分解代谢过程以及参与有丝分裂细胞周期的泛素蛋白连接酶活性调控。 细胞构成(CC)主要集中在纺锤体微管、亚体、胞质、纺锤体、浓缩核染色体外着丝粒、细胞表面、微管细胞骨架、核质、减数分裂纺锤体和着丝粒。 分子功能(MF)与蛋白激酶结合、 磷酸蛋白磷酸酶活性和蛋白结合方面有关。 KEGG 主要富集在孕激素介导的卵母细胞成熟、细胞周期、卵母细胞减数分裂和FoxO 信号等通路,见图5e。

图5 枢纽基因的共表达网络及功能富集分析
2.7 HCC 患者中枢纽基因的免疫细胞浸润分析利用TIMER 数据库评估了CCNB1、ASPM 和AURKA 与免疫细胞浸润之间的关系。 如图6a 所示,CCNB1 表达与浸润性B 细胞、巨噬细胞、中性粒细胞和树突状细胞呈显著正相关。ASPM 表达与浸润性B 细胞、CD4+T 细胞、巨噬细胞、中性粒细胞和树突状细胞呈显著正相关(P<0.05),见图6b。AURKA表达与浸润性B 细胞和树突状细胞呈显著正相关(P<0.05),见图6c。
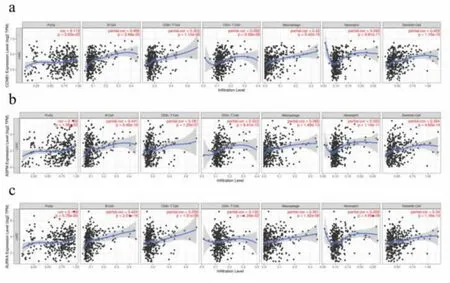
图6 不同表达的枢纽基因与免疫细胞浸润之间的相关性
3 讨论
本研究的结果表明CCNB1、ASPM 和AURKA在HCC 中高表达且与HCC 患者不良的OS 和DFS 相关。 目前,有许多证据表明这3 个枢纽基因在HCC 的发生发展中具有重要作用。有研究发现,在HCC 患者中AURKA 的表达与肿瘤大小、OS 和DFS 长短密切相关[12-13]。 也有研究表明CCNB1、ASPM 和AURKA 在HCC 中过度表达, 可能是潜在的HCC 早期诊断生物标志物[14-17]。 最近的一项研究显示,CCNB1 基因的敲除可显著抑制HCC 中的细胞增殖、迁移和侵袭[18]。 本研究对于CCNB1、ASPM 和AURKA 在HCC 中的分析结果与之前的研究所报道的一致[19-21]。 因此,这3 个基因具有作为HCC 预后标志物的潜力。然而,当前还没有相关研究阐明其在HCC 发生、 发展以及转移中的具体作用机制。
有研究表明,CCNB1、ASPM 和AURKA 是 参与细胞分裂和增殖的重要因素[22-24]。 一些研究还表明, 这3 个枢纽基因在其他疾病中的机制主要归因于其对多种信号通路的调节[25-27]。 本研究探索了20 个可能与CCNB1、ASPM 和AURKA 功能相关的基因,其中一些已被确定为其重要的调节因子。据Krause 等[28]报道,PLK1 与肿瘤蛋白p53(TP53)的DNA 结合域结合以阻断其转录活性可上调CCNB1 的表达。 此外,TPX2 是一种重要的辅助因子, 参与将AURKA 定位到纺锤微管并直接激活AURKA[29]。 有研究表示,AURKA 通过调节HCC 的上皮—间质转化和肿瘤干细胞特性促进肿瘤转移,这不利于HCC 患者的预后[30]。 我们的研究进一步对这3 个枢纽基因进行功能富集分析, 以了解其在HCC 中的生物学功能。结果表明这3 个枢纽基因主要参与PID-PLK1 通路、G2/M 转换和有丝分裂的细胞周期蛋白A/B1/B2 的相关调节。CCNB1是细胞周期中的关键因子, 由STAT3 通过E2F 调节G2/M 期检查点进行调节[31]。
近年来,大量研究表明,系统评价肿瘤浸润免疫细胞对预测临床预后和发展免疫治疗具有重要意义[32]。Nataliya 等[33]应用CIBERSORT 评估健康人肝脏与HCC 或其邻近组织中免疫细胞的相对比例, 发现HCC 中未活化的肥大细胞、 总B 细胞、CD4+和CD8+T 细胞均增加,而与健康肝脏相比,活化的肥大细胞、单核细胞和浆细胞减少。 目前,很少有研究表明肿瘤相关巨噬细胞、自然杀伤细胞、树突状细胞、TIM3、PD-L1、PD-L2 等在HCC 患者预后中的重要性[34-37]。 因此,我们分析了B 细胞、CD8+T 细胞、CD4+T 细胞、巨噬细胞、中性粒细胞和树突状细胞在CCNB1、ASPM 和AURKA 不同表达水平下的浸润率并且观察到它们的表达水平均与这些免疫细胞浸润有关, 这对进一步了解HCC 的免疫状况有一定的帮助。
本研究仍存在几个局限性。 首先,我们使用了来自多个不同数据库的数据, 很难确保不同数据库之间的一致性。 其次,未对这些数据库中的数据进行验证。 最后,没有进一步研究和验证这3 个枢纽基因在HCC 患者中的确切机制。
综上所述,我们的研究表明CCNB1、ASPM 和AURKA 的高表达与HCC 的预后和免疫微环境显著相关。 因此,我们希望本研究能够有助于临床医生为肝癌患者选择合适的预后标志物。
