OPTN突变致RGCs自噬异常引起POAG的研究进展
2022-06-28邓娅青江海波
邓娅青, 江海波
(中南大学湘雅医院眼科中心,湖南省长沙市 410008)
青光眼是临床常见的不可逆性致盲性眼病,是导致双眼失明的主要原因之一,其特征是视网膜神经节细胞(retinal ganglion cells,RGCs)退行性变和视神经损伤。原发性开角型青光眼(primary open angle glaucoma,POAG)为其主要类型,POAG的主要危险因素包括高眼压、高龄、女性以及家族遗传史等[1-2]。根据眼内压,POAG可分为高眼压型青光眼(眼压>21 mmHg)和正常眼压型青光眼(normal tension glaucoma,NTG)(眼压≤21 mmHg),NTG占POAG的比例高达1/3[3]。POAG具有遗传异质性,与OPTN基因突变联系密切[4]。
1 OPTN突变与POAG
2002年,Rezaie等[5]学者首次证明OPTN突变与成年发病型POAG相关,且可见于16.7%的遗传性NTG患者中。随后研究者发现在NTG的致病因素中,OPTN突变可占1.5%[6]。OPTN基因突变类型包括同义突变和错义突变,发生错义突变时Optineurin原有的正常结构和功能将受到影响。目前发现与POAG相关的错义突变有H26D、E50K、M98K、E103D、691_692insAG(OPTN第691和第692位碱基中间插入了AG两个碱基)、T202R、A336G、A377T、H486R、R545Q等(表1)[3,7-9]。E50K、M98K、691_692insAG、R545Q是最先从54个POAG家族中检测到的OPTN突变,这些家族中大部分人眼压正常[3],其中E50K与NTG的相关性最强,存在于<2%的NTG和<1%的POAG患者中[6]。E50K突变和M98K突变会导致RGCs自噬功能异常。E50K突变最早在高加索和西班牙人群中发现,患者在眼压较低时即出现早发性严重视神经损伤,与无E50K突变者相比,E50K突变者眼压更低、杯盘比更大、视野损失更严重[10]。M98K突变与其他突变类型不同的是,它引起的青光眼具有种族特异性[11],仅与亚洲人群青光眼相关,而与高加索人群青光眼不相关。M98K突变不会直接导致NTG,但它可能会增加某些人群患青光眼的风险[10]。

表1 与POAG相关的OPTN突变及Optineurin变化
2 OPTN编码Optineurin
人类OPTN基因(OMIM-602432)位于10号染色体短臂1区3带(10p13),跨越大约37 kb的基因组区域,由3个非编码外显子和13个编码外显子组成[7],其编码的蛋白质Optineurin由577个氨基酸组成,相对分子质量为74 kDa[9]。OPTN突变会导致Optineurin功能异常,与多种蛋白的相互作用发生改变,进而引发一系列疾病,其中POAG和肌萎缩性脊髓侧索硬化症最为常见[8]。OPTN突变导致的其他疾病还有阿尔茨海默病、帕金森病和佩吉特氏骨病[12]。
2.1 Optineurin的结构
Optineurin是一种不具有任何酶活性的螺旋蛋白。它包含一个泛素结合结构域(ubiquitin-binding domain,UBD)、一个锌指结构域(Zinc finger,ZF)、至少一个亮氨酸拉链(leucine zipper,LZ)、一个微管相关蛋白1轻链3结合结构域(LC3-interaction region,LIR)、多个卷曲螺旋状结构域(coiled coil domains)[9],见图1。其N端与TBK1、RAB8、MYPT1、LC3、mGluR1等蛋白相互作用,C端与Htt、RIP1、Myosin VI、CylD等蛋白相互作用[13]。
Optineurin在人体大脑、肝脏、心脏、视网膜、眼球前房的小梁网、睫状体的非色素纤毛上皮中都有表达,在RGCs中表达水平较高[10]。在亚细胞水平,Optineurin主要位于细胞质、核周区域、高尔基体和跨高尔基网络区域[10]。内源性Optineurin是一种寿命较短的蛋白质,半衰期约为8 h,在正常的体内平衡状态下,内源性Optineurin主要通过泛素蛋白酶体系统降解[7]。
2.2 Optineurin的功能
Optineurin通过与多种蛋白质如Rab8、Myosin VI、Huntigtin、TBC1D17、TFRC等相互作用,具有参与自噬、囊泡运输、调节细胞死亡、有丝分裂、细胞保护、抗病毒等功能[9]。自噬是细胞内一种参与维持细胞内环境稳态的程序性分解代谢过程,广泛存在于各种真核细胞中,和泛素蛋白酶体系统共同成为细胞内两种主要的蛋白质降解途径[14]。自噬可调节凋亡、炎症和适应性免疫,很多情况下在细胞中起保护作用,但自噬的过度激活对细胞有害,可能导致细胞死亡[15-16]。自噬功能异常与一些神经退行性疾病的发生、发展有关,如阿尔茨海默病和青光眼[17]。
Optineurin作为一种选择性自噬受体,在选择和招募需要被降解的蛋白质和细胞器中起着关键作用[18]。它通过UBD结构域与泛素化的蛋白质或细胞器结合,通过LIR结构域招募LC3,形成自噬小体包裹需要被降解的蛋白质或细胞器(图2)[19]。Optineurin与LC3的相互作用是通过TBK1来调控的,TBK1是一种丝氨酸激酶,使Optineurin第177位的丝氨酸(位于LIR结构域)磷酸化,从而增强Optineurin与LC3的相互作用[4]。

图1 Optineurin结构以及其与POAG相关的突变类型示意图
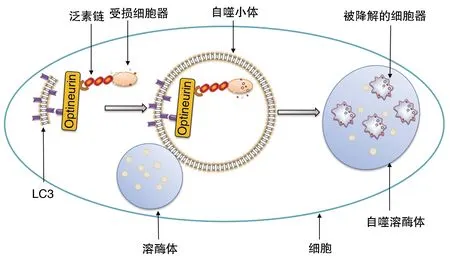
图2 Optineurin参与自噬过程示意图
3 RGCs自噬与POAG
RGCs是位于视网膜最终段的神经细胞,其轴突为视神经纤维,分布于视网膜表面,汇聚于视乳头,穿出筛板后形成视神经。RGCs的进行性死亡是POAG最重要的病理学表现,但其具体死亡机制目前仍不明确[20]。POAG中RGCs进行性死亡的可能机制是凋亡,但近年来研究发现自噬也参与这个过程[21]。研究发现,自噬可介导慢性高眼压引起的RGCs死亡,从而参与POAG的发病机制[22-23]。RGCs自噬可由缺血、缺氧、兴奋性氨基酸增多等应激状态激活,正常情况下对RGCs起保护作用,但当应激达到一定阈值时凋亡被激活,RGCs发生死亡[24]。除RGCs自噬之外,小梁网细胞自噬、神经胶质细胞自噬等也参与POAG的发病机制[25]。
4 不同OPTN突变类型致RGCs自噬异常
4.1 E50K突变
2014年Chalasani等[26]发现,表达E50K突变蛋白的RGC-5中自噬小体数量增多而自噬溶酶体数量减少,认为E50K突变蛋白可能使RGC-5自噬过程受阻而发生死亡,且自噬受阻可能发生在自噬小体形成之后。用雷帕霉素(自噬诱导剂)处理RGC-5可显著抑制E50K突变蛋白诱导的RGC-5死亡。E50K突变致RGCs自噬功能受阻与其和TBK1、Rab8、TFRC等在自噬过程中起重要作用的蛋白相互作用异常有关[8]。Ying等[27]得出E50K突变蛋白可以诱导大鼠RGCs自噬增强。同时VanderWall等[28]用胚胎干细胞系模型证明E50K突变的RGCs自噬过程受阻。
4.2 M98K突变
M98K突变致青光眼的主要机制为增强细胞内自噬,导致TFRC降解过多以及RGCs凋亡过度[11]。Sirohi等[11]在RGC-5中表达M98K突变蛋白,发现与没有表达M98K突变蛋白的RGC-5相比,前者诱导更多的自噬小体形成,这与M98K突变蛋白S177(第177位丝氨酸)磷酸化增强有关,S177磷酸化增强可使M98K突变蛋白与LC3相互作用增强,促进自噬小体的形成。M98K突变蛋白S177磷酸化增强是由TBK1调节的,M98K突变蛋白表达后激活TBK1,两者相互作用。在RGC-5中过表达M98K突变会导致自噬体和自溶酶体数量增加,自噬增强,使用自噬抑制剂阻断TFRC降解、过表达TFRC或者在细胞培基中加入铁离子,都可减少M98K突变诱导的细胞死亡[13]。M98K突变蛋白诱导的自噬增强也可能依赖于Rab12 GTP酶[12]。M98K突变蛋白过度激活自噬也可能与其和泛素、Rab12间的相互作用出现异常有关[13]。
5 总结与展望
自噬作为一种潜在的治疗靶点,已经在很多领域引起关注,尤其在神经退行性疾病相关领域[29]。E50K和M98K突变致RGCs自噬功能异常引起RGCs死亡在POAG的发病机制中起重要作用,因此调节自噬功能相关药物可能具有预防和治疗POAG相关RGCs死亡的潜力[8]。TBK1蛋白激酶抑制剂、TBC1D17、雷帕霉素等已被证明具有调节自噬的功能,可能会减少自噬功能异常导致的RGCs死亡[8],它们能否成为一种新型的抗青光眼药物,尤其在治疗正常眼压型青光眼方面的价值,还值得进一步研究。
