CuZn 溶胶中不同溶剂对CuZnAl 浆状催化剂催化合成气制乙醇性能影响
2022-06-27顾尹婷黄金花
顾尹婷,韩 春,黄金花,黄 伟
(太原理工大学 省部共建煤基能源清洁与高效利用国家重点实验室,山西 太原 030024)
随着能源需求的增加、原油储量的减少和环境问题的加剧,各国深入研究了不同原料路线的化学品合成方法。煤、天然气和生物质等一次能源可通过气化或重整技术得到合成气(H2和CO的混合物)。合成气作为化工合成中的重要平台[1],可经不同的工艺路线生产各种化学品,如乙醇、乙酸和甲醇等。乙醇可以用作清洁替代燃料和燃料添加剂,还可作为合成药物、表面活性剂和聚酯等精细化学品的原料[2]。合成气制乙醇中,对乙醇或低碳醇具有选择性的催化剂主要有铑基催化剂[3-4]、钼基催化剂[5-7]、改性费托合成催化剂[8-10]和改性铜基甲醇合成催化剂[2,11-12]。铑基催化剂C2产品选择性高,但催化剂价格昂贵;钼基催化剂耐硫,但制备条件苛刻;改性的铜基催化剂产物主要是甲醇和异丁醇,乙醇较少。
研究者[2,13-16]通常认为乙醇等C2+醇合成需要两种活性位点,一种负责CO解离,形成表面CHx物种;另一种负责CO插入,形成C—O键。本课题组前期工作发现,采用所研发的完全液相法制备的CuZnAlOOH浆状催化剂,在不添加助剂时就具有较好的乙醇合成能力[17-18]。乙醇合成对催化剂结构敏感,高性能乙醇催化剂的重现性和稳定性不佳。理论计算表明,在CuZnAlOOH催化剂上,乙醇合成有利的活性位点为Cuδ+(0 <δ< 1)[19]和Cu/γ-AlOOH(001)[20],其中具有0.43 和0.51 中等价态Cuδ+的亚稳态活性位结构有利于乙醇生成,但在反应气氛下可能发生价态改变,偏离中等价态,成为甲醇合成占优势的活性位点,从而使乙醇合成的稳定性下降。甲醇合成反应的活性中心通常认为是铜锌合金位点[21],Cu0价态的(111)[22-23]、(211)[24]、(110)[23,25]和(100)[12,23]表 面。与 乙 醇 合成位点相比,在反应气氛下甲醇活性位点稳定性更强。
为解决上述问题,本文在完全液相法基础上,结合高总醇选择性的CuZn溶液法与高乙醇占比的CuZn溶胶法[26-27],将催化剂前驱体制备所需的铜盐、锌盐等分成两份,分别制成CuZn溶胶与CuZn溶液,依次添加制作催化剂,进而研究CuZn溶胶中溶剂变化对CuZnAlOOH浆状催化剂性能的影响。结合催化剂性能评价结果,通过多种表征手段获得催化剂反应前后结构变化信息,关联催化剂构效关系,探讨溶剂影响的原因。
1 实验部分
1.1 实验原料与试剂
化学试剂与原料如表1 所示。

表1 化学试剂与原料Table 1 Chemical reagents and materials
1.2 催化剂制备
(1)溶液配制:将0.1250 mol Cu(NO3)2·3H2O与0.0625 mol Zn(NO3)2·6H2O溶于乙二醇中,使用磁力搅拌器搅拌12.0 h得到Cu2+浓度为5.00 mol/L的溶液A(即CuZn溶液)。将0.1250 mol Cu(NO3)2·3H2O与0.0625 mol Zn(NO3)2·6H2O溶于蒸馏水(或乙二醇、乙醇)中,搅拌12.0 h得到Cu2+浓度为5.00 mol/L的溶液B。将0.050 mol柠檬酸(CA)溶于蒸馏水(或乙二醇、乙醇)中,搅拌12.0 h得到CA浓度为1.67 mol/L的溶液C。将20.424 g异丙醇铝充分研磨后溶于40 mL乙二醇中,搅拌12.0 h得到溶液D。
(2)催化剂前驱体制备:将溶液B与溶液C混合于500 mL三口烧瓶内,在80 °C水浴中以300 r/min搅拌成胶,得到CuZn溶胶。在1000 mL三口烧瓶内加入0.450 g PVP与70 mL蒸馏水,在80 °C水浴以300 r/min搅拌。待PVP溶解后,在5 min内将溶液D全部滴入,开盖静置1.0 h后重新开启搅拌,加适量浓硝酸调节pH为3,得到铝溶胶。0.5 h后在水浴温度95 °C条件下滴加CuZn溶液,1.0 h后滴加CuZn溶胶,滴加结束后回流搅拌10.0 h,取出在室温下冷却,静置老化10 d,得到催化剂前驱体。
(3)成品浆状催化剂制备:老化结束后向前驱体中加入300 mL液体石蜡和0.5 mL Span-80,将混合物倒入1000 mL三口烧瓶,将三口烧瓶置于智能控温加热套内搅拌升温至280 °C,在280 °C维持8.0 h,得成品浆状催化剂。
当以水作为CuZn溶胶中的溶剂(即溶液B与溶液C中使用水作为溶剂),制得的催化剂记为“Cat.w”,以乙二醇为溶剂的记为“Cat.eg”,以乙醇为溶剂的记为“Cat.et”。以Cat.w催化剂为例,制备流程如图1 所示。
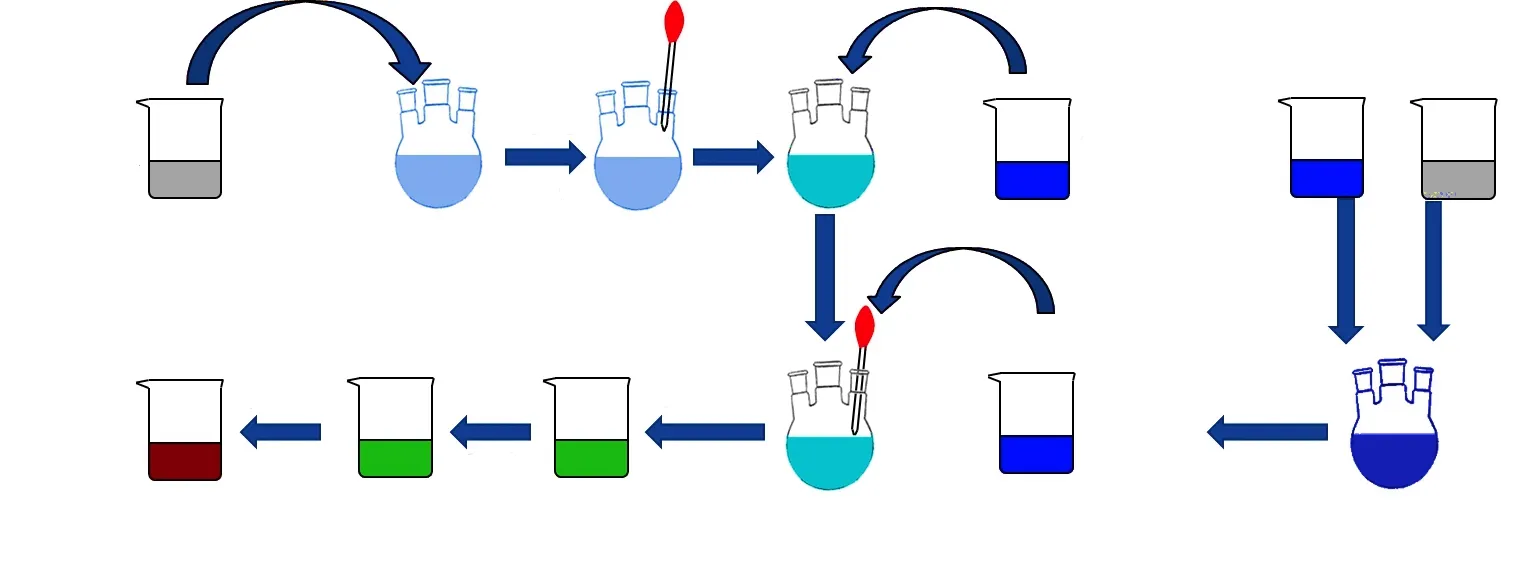
图1 催化剂Cat.w制备流程Fig. 1 Catalyst Cat.w preparation process
1.3 催化剂性能评价
催化剂性能评价在500 mL加压搅拌式模拟浆态床反应器中进行,催化剂床固含率为11%。反应前催化剂在反应釜内在线常压程序升温还原,还原气(n(H2)/n(N2)=1/4)流量为75 mL/min,还原过程中搅拌器转速为1000 r/min,升温速率为0.5 °C/min,还原温度为280 °C,在该温度下保持600 min。催化剂在线还原后在60 °C以下用H2加压至4.5 MPa,开启反应气(n(H2)/n(CO) = 2/1),流量为150 mL/min,以升温速率0.5 °C/min升至反应温度250 °C进行反应,反应时搅拌器转速1000 r/min,每种催化剂的评价时间至少4 d。
反应产物在GC-950 气相色谱上进行离线分析。该色谱配有两个氢火焰检测器(FID)、一个热导检测器(TCD)。进入色谱的样品分为两路,一路经GDX-502 色谱柱分离后通过FID对甲醇(MeOH)、乙 醇(EtOH)、C1~C5烃 类 和 二 甲 醚(DME)进行检测;另一路经TDX-01 色谱柱分离后先通过TCD检测器检测H2,之后通过镍转化炉进入FID,检测CO和CO2。气相产物每8.0 h取样1 次,每天采样3 次,将3 次采样数据的平均值作为该日气相产物分析结果。液相产物每日采集1 次。综合1天内气、液产物的分析结果给出当天的催化剂性能数据。定量方法采用甲烷归一化法,以各通道中甲烷含量为基准对其余各组分含量进行定量计算,经碳平衡后计算各产物的选择性和CO转化率。
CO的转化率计算公式为:

式中,vi为组分i分子内碳原子个数;yi为组分i物质的量分数;下标i代表某组分。
各产物选择性计算公式为:

总醇选择性计算公式为:

甲醇(乙醇)占总醇选择性计算公式为:

1.4 催化剂表征
反应前后的浆状催化剂在80 °C下使用高沸程(60~90 °C)石油醚进行索氏抽提48.0 h,去除催化剂表面的液体石蜡,烘干后得到粉末状催化剂样品用以进行各项表征。
氢气程序升温还原(H2-TPR)表征在天津先权仪器厂生产的TP-5080 多用吸附仪上完成。催化剂用量为50 mg,催化剂在40 mL/min氢氮混合气(5%H2/95%N2)中以升温速率10 °C/min从50 °C升温至450 °C,热导池检测尾气中的H2含量。
X射线光电子能谱(XPS)表征采用Thermo Fisher公司的Thermo Scientific K-Alpha+能谱仪,单色化Al Kα射线(hv= 146.6 eV),束斑400 μm,分析室真空度为5 × 10-7Pa,各元素结合能以标准C 1s的结合能(284.8 eV)校准。
高分辨透射电子显微镜(HRTEM)表征采用日本JEOL公司的JEM-2100F场发射透射电镜,操作电压200 kV。
N2物理吸/脱附分析在美国康塔公司的Quanta chrome QDS-30 物理吸/脱附仪上进行。测试前催化剂样品先在200 °C下脱气 4.0 h,之后在-196 °C下测定吸/脱附曲线,以BET公式计算比表面积,BJH法计算孔径分布。
O2程序升温氧化-质谱联用(O2-TPO-MS)表征在天津先权仪器厂生产、配备质谱检测器(QIC-20)的TP-5080多用吸附仪上完成。催化剂用量为50 mg,催化剂先在氢氮混合气(5%H2/95%N2)中,150 °C下吹扫0.5 h,降温至50 °C下吹扫25 min,再用氧氩混合气(5%O2/95%Ar)进行程序升温氧化,以10 °C/min的升温速率从50 °C升温到1000 °C,以质荷比44 的碎片标记氧化产物CO2。
2 结果与讨论
2.1 催化剂性能测试
2.1.1 CO转化率、产物选择性与醇分布
催化剂的CO转化率、产物选择性及醇分布随反应时间的变化如图2 所示。
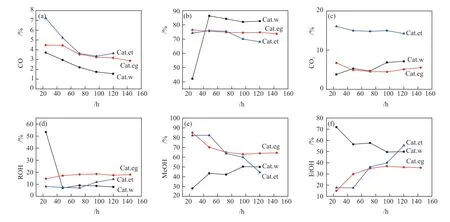
图2 CO转化率(a)、烃类选择性(b)、CO2 选择性(c)、总醇选择性(d)、甲醇占总醇选择性(e)和乙醇占总醇选择性(f)随反应时间的变化Fig. 2 Changes of CO conversion (a), hydrocarbon selectivity (b), CO2 selectivity (c), ROH selectivity (d), MeOH selectivity in ROH (e) and EtOH selectivity in ROH (f) with reaction time
由于反应初期催化剂存在诱导期[28-29],各性能指标波动较大,部分催化剂的个别指标在评价期间(一般120.0 h)未达到平稳,因此本文以每种催化剂120.0 h(Cat.eg为144.0 h)的性能值为衡量标准。由图2 可知,以水为溶剂的Cat.w催化剂转化率与总醇选择性最低,副产物烃类选择性最高,达80%以上,但乙醇占比较高,达50%。以乙二醇为溶剂的Cat.eg催化剂,CO转化率在144.0 h的反应时间内呈下降趋势,总醇选择性最高,但乙醇占比和CO2选择性最低。以乙醇为溶剂的Cat.et催化剂,CO转化率在72.0 h后实现平稳,烃和CO2选择性随反应时间增加缓慢下降,总醇选择性缓慢上升,乙醇占比随反应时间增加快速上升,从24.0 h时不足20%上升到120.0 h时接近60%,相应的甲醇占比快速下降。综上,催化剂Cat.et总体指标随反应时间增加变佳,具有应用前景。与本课题组研制的其他同类催化剂及文献报道相比[18],本文所制催化剂醇产品中只有甲醇和乙醇,后续分离负担较轻,是其优势所在。
2.1.2 烃类分布与碳链增长机制
烃类分布随时间的变化及A-S-F分布相关性分析如图3 所示。理论计算表明[30-33],铜基催化剂上烃类产物与乙醇均来源于CHx物种的进一步反应,加氢、耦联分别得到甲烷和乙烷,CO插入则得到乙醇,而甲醇是通过CO直接加氢得到。因此,高乙醇占比催化剂均伴随着高的烃选择性,进而导致总醇选择性不高[7-9],这也是合成气制低碳醇的共性难题。在这个机理下,烷烃与乙醇共用一个中间体,而甲醇与其为平行反应。将产物烃类分布用A-S-F法进行相关性分析发现(图3(d)~(f)),3 种催化剂均不十分符合A-S-F分布,表明其碳链增长并非一种机理所定,即存在不同的活性位点。BAI等[34]研究表明,完全液相法制备的CuZnAl有两种路径实现碳链增长,一种是在CuZn界面CO和H2生成甲醇,甲醇在CuAl界面解离生成CH3表面物种,之后CO插入CH3生成CH3CO物种,再持续加氢生成乙醇,速控步骤为CH3的生成;另一种是CO和H2在CuZn界面生成CHO并偶联为OHC—CHO,然后持续加氢生成乙醇,速控步骤为CHO的生成。
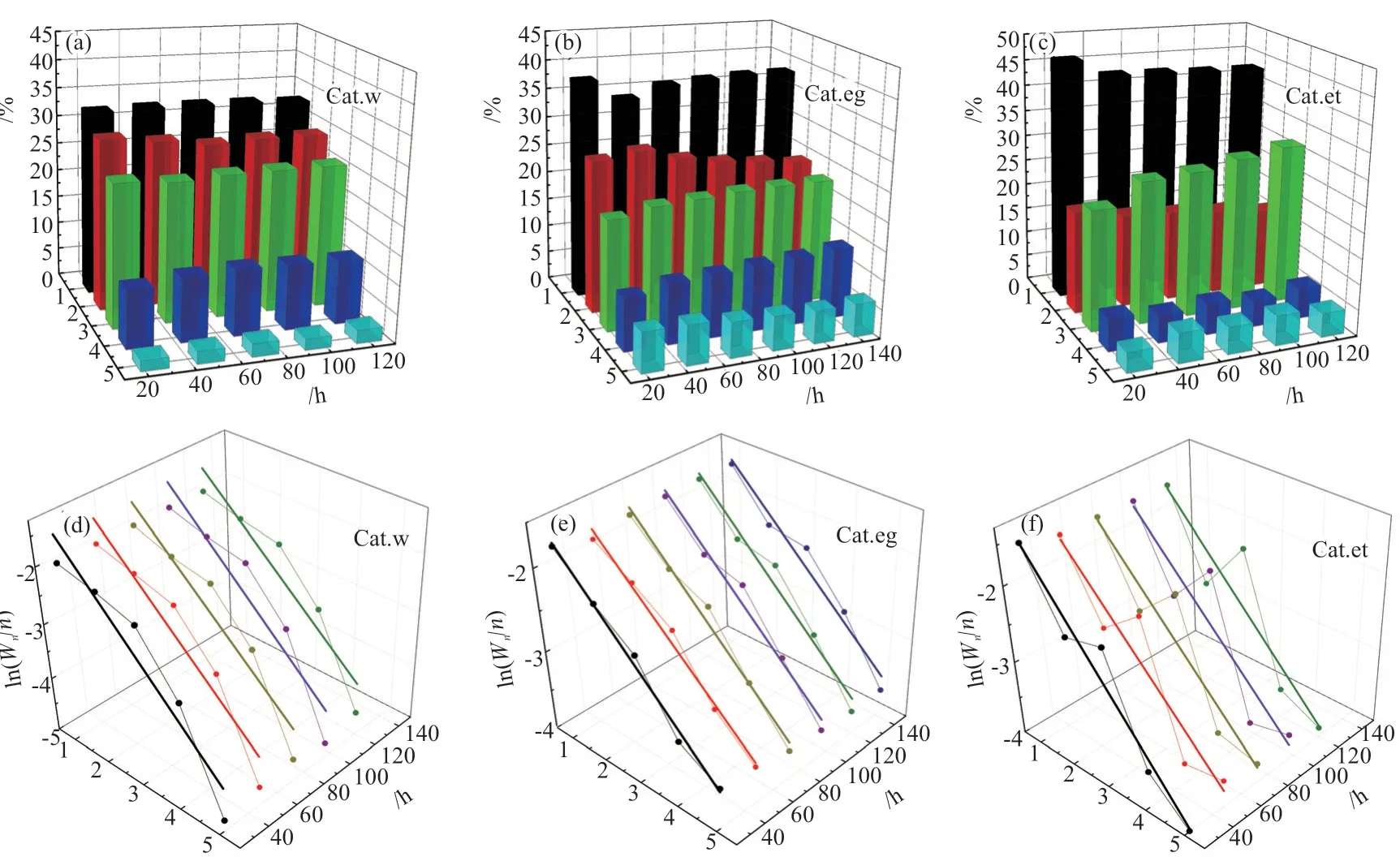
图3 不同碳数烷烃占总烷烃的比例以及A-S-F分布随时间的变化Fig. 3 Changes of proportion of hydrocarbons with different carbon numbers in total alkanes and A-S-F distribution with time
本文催化剂制备是将Cu、Zn分成两部分,一部分制成CuZn溶胶,另一部分制成CuZn溶液然后与Al溶胶混合,再加入CuZn溶胶,目的是调控Cu0和Cu+的比例。然而,这种方法也导致了催化剂中存在CuZn和CuZnAl两种结构,确保了上述两种路径并存的可行性。从高乙醇占比催化剂均伴随着高的烃选择性实验结果看,本文催化剂主要以第一条路径生成乙醇。Cat.et催化剂中(图3(f)),乙烷存在明显下降,丙烷存在明显上升,且随着反应时间与A-S-F分布的偏差越来越大,而乙醇占比呈向上趋势(图2(f)),印证了乙醇的生成是CO插入CH3的结论。Cat.eg催化剂总体呈现了与Cat.et相似的结果,而Cat.w呈现了较多的C4烃和较少的C5烃的生成,显示出与其他两种催化剂的明显区别,相关原因还需进一步探讨。
表2给出了各催化剂A-S-F拟合的相关系数R2值。由表2 可知,Cat.et催化剂乙醇占总醇选择性最高,R2值最小,偏离A-S-F更远,而Cat.eg反之。这可作为存在CHO耦合碳链增长的证据。
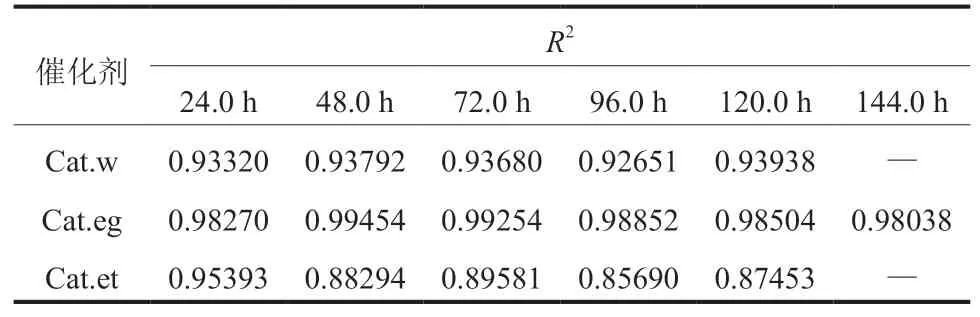
表2 A-S-F线性拟合的R2 随时间的变化Table 2 Changes of R2 of A-S-F linear fitting with time
2.2 XRD表征
图4为反应前后不同催化剂XRD表征谱图。由图4 可知,反应前后催化剂XRD谱图非常相近,没有明显的变化。Cu主要以Cu0的形式存在,不存在CuO相,这是完全液相法制备催化剂的特征之一,反应前Cu2+已被还原为低价铜[35]。在衍射角37°附近可以隐约观察到Cu2O衍射峰,没有发现任何明显的Zn、ZnO或含Al物相的衍射峰,说明催化剂中Zn、Al物种没有达到XRD可探测的聚集态或为无定型态[36-37]。23.0°可归属为无定形碳的衍射峰,反应后该峰强度有所下降,表明反应后催化剂表面碳含量有所下降[18]。
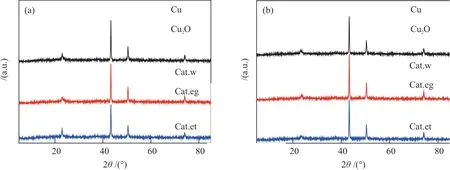
图4 反应前(a)和反应后(b)催化剂的XRD谱图Fig. 4 XRD spectra of catalysts before (a) and after (b) reaction
通过谢乐公式计算了催化剂反应前后不同晶面方向上的晶粒尺寸,结果列于表3。由表3 可知,3 种催化剂反应后在垂直于Cu(111)方向上的尺寸增长最小或呈负增长。其中,Cat.w催化剂在(200)晶面方向上反应后尺寸增长18%,而(220)晶面上仅增长5.9%;Cat.eg在Cu(200)晶面方向上的晶粒尺寸较反应前仅增长3.3%,但在(220)晶面增长了43.7%;Cat.et在(200)与(220)方向上的尺寸增量差距较小,分别为15.0%和21.6%。显然,不同溶剂催化剂反应过程中均出现了Cu原子重排现象,并且长成了不同的Cu0晶粒形貌。这种变化是不同溶剂催化剂中Cu所处的化学环境与反应环境共同作用的结果。Cat.et的Cu0晶粒形貌变化不大,说明反应过程中其结构更稳定。
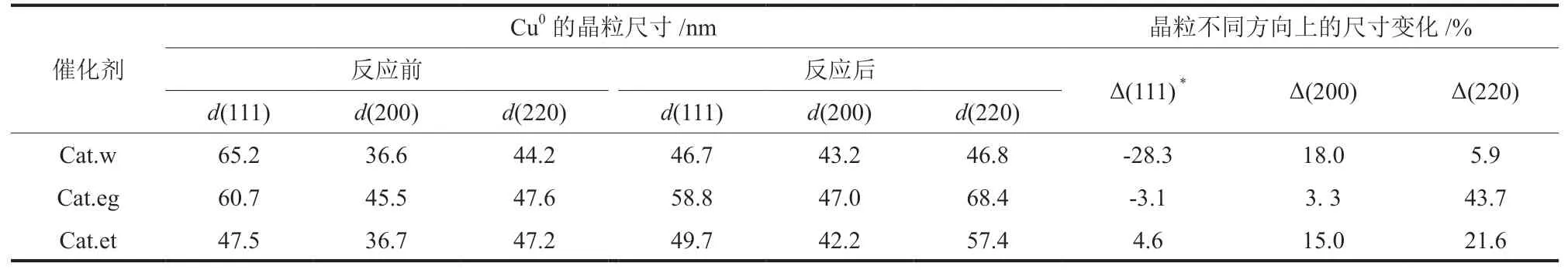
表3 反应前后催化剂Cu0 各晶面方向上的晶粒尺寸及其变化量Table 3 Cu0 crystallite size in each direction and its change of catalysts before and after reaction
2.3 H2-TPR表征
图5为催化剂反应前后的H2-TPR谱图。由于完全液相法所制催化剂中,Al以AlOOH的形式存在,AlOOH属于介稳结构,尽管已经证明其在液体石蜡和在本文的反应条件下稳定[18],但还是对AlOOH开展了H2-TPR表征(图5(c)),发现AlOOH的H2-TPR曲线在约200 °C以后开始缓慢下移,下移的原因可能是AlOOH在200 °C以后发生分解。无论反应前还是反应后,催化剂的H2-TPR谱图均存在3 个还原峰,分别记为ɑ、β和γ。其中ɑ为易还原Cu+的低温还原峰,温度范围在180~260 °C[38-40],该峰较为平缓,说明易还原的Cu+含量极少;β为与Zn、Al存在强相互作用的Cu+物种的还原峰,温度范围280~360 °C,文献报道该Cu+物种对乙醇的生成有利[41-42];γ应为催化剂表面碳物种的加氢脱附峰,反应后该峰明显变小,表明反应后催化剂表面碳物种数量减少,这与XRD中归属于无定型碳的衍射峰强度在反应后降低一致。

图5 反应前(a)和反应后(b)催化剂及拟薄水铝石AlOOH (c)的H2-TPR谱图Fig. 5 H2-TPR spectra of catalysts before (a) and after (b) reaction and AlOOH (c)
根据谱图中各还原峰的面积,扣除AlOOH对基线的影响,计算了反应前后催化剂还原的耗氢量,见表4。由表4 可知,在反应后的样品中,溶胶溶剂为乙醇的催化剂总耗氢量最大,水的次之,乙二醇的总耗氢量最少,该结果与乙醇占比顺序基本一致。可见,催化剂中高Cu+含量(难还原Cu+与易还原Cu+含量之和)是合成乙醇的关键要素。采用本文的制备方法,通过改变CuZn溶胶中的溶剂,能有效地实现对催化剂中Cu+含量的调控,进而获得高乙醇合成的催化剂。
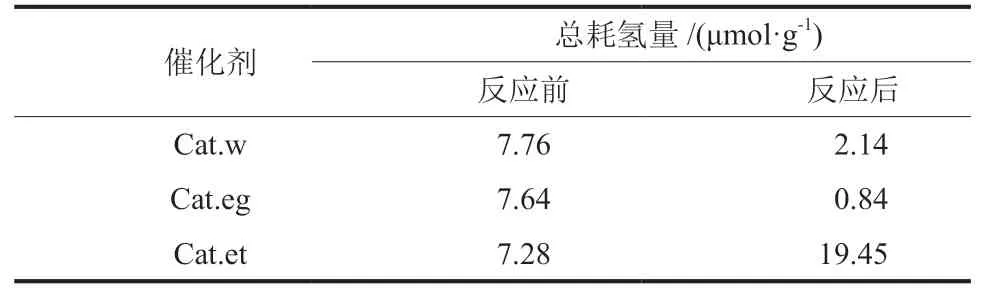
表4 反应前后催化剂的总耗氢量Table 4 Total H2 consumption of catalysts before and after reaction
2.4 XPS及XAES表征
图6为不同溶剂催化剂反应前后Cu 2p、Zn 2p和Al 2p XPS表征谱图。由图6 可知,Al 2p峰型反应前后均对称,位于77.0 eV左右肩峰为Cu 3p峰。反应前后Al 2p结合能变化不大,约为74.5 eV,属于AlOOH中的AlO6八面体[43]。Cu 2p谱图中没有明显的卫星峰,说明催化剂表面基本不存在CuO。

图6 反应前(a) 、(c)、 (e)和反应后(b) 、(d) 、(f)催化剂的XPS谱图Fig. 6 XPS spectra of catalysts before (a), (c), (e) and after reaction (b), (d), (f)
基于Cu的俄歇谱,可以区分Cu0和Cu+,见图7和表5。由表5 可知,反应后n(Cu0)/n[(Cu++Cu0)]普遍较反应前降低,表明Cu+相对量增加,且催化剂Cat.et和Cat.w反应后n(Cu0)/n[(Cu++Cu0)]相近,这也与反应120.0 h两种催化剂的乙醇占比接近相一致。进一步表明高的Cu+相对量有利于乙醇的生成。

图7 反应前(a)和反应后(b)催化剂的Cu LMM俄歇谱Fig. 7 Cu LMM spectra of catalysts before (a) and after (b) reaction
图8为反应前后催化剂Zn俄歇电子能谱及其分峰结果,相关参数列于表6。由图8 和表6 可知,催化剂表面的Zn物种主要为Zn(2-δ)和Zn2+,二者的俄歇电子动能分别约为990.1 eV和987.1 eV。在反应前各催化剂的n(Zn(2-δ))/n(Zn2+)在0.371~0.416 之间,反应后在0.332~0.461 之间,分布范围变宽。反应前后n(Zn(2-δ))/n(Zn2+)最低的为Cat.et催化剂,最高的为Cat.eg。根据文献[44-45]报道,Zn氧空位缺陷Zn(2-δ)可以为H2的解离和CO/CO2的吸附提供活性位点,促进CO/CO2加氢生成CHO,进而生成甲醇。前文已经提到,碳链增长的一个路径是通过合成气生成甲醇再解离生成CH3,然后CO插入生成CH3CO,继而加氢生成乙醇。但Cat.eg缺乏高的表面Cu+含量(见表5 中反应后相应值),对甲醇解离生成CH3这一速控步骤缺乏推动作用,故其甲醇占总醇比例最高,乙醇占比最低。显然,Zn(2-δ)不能单独产生作用影响乙醇合成,而是与Cu、Al协同最终生成乙醇。

表6 反应前后催化剂表面Zn(2-δ)与Zn2+俄歇电子动能及其含量之比Table 6 Auger electron kinetic energy and content ratio of surface Zn(2-δ) and Zn2+ in catalysts before and after reaction
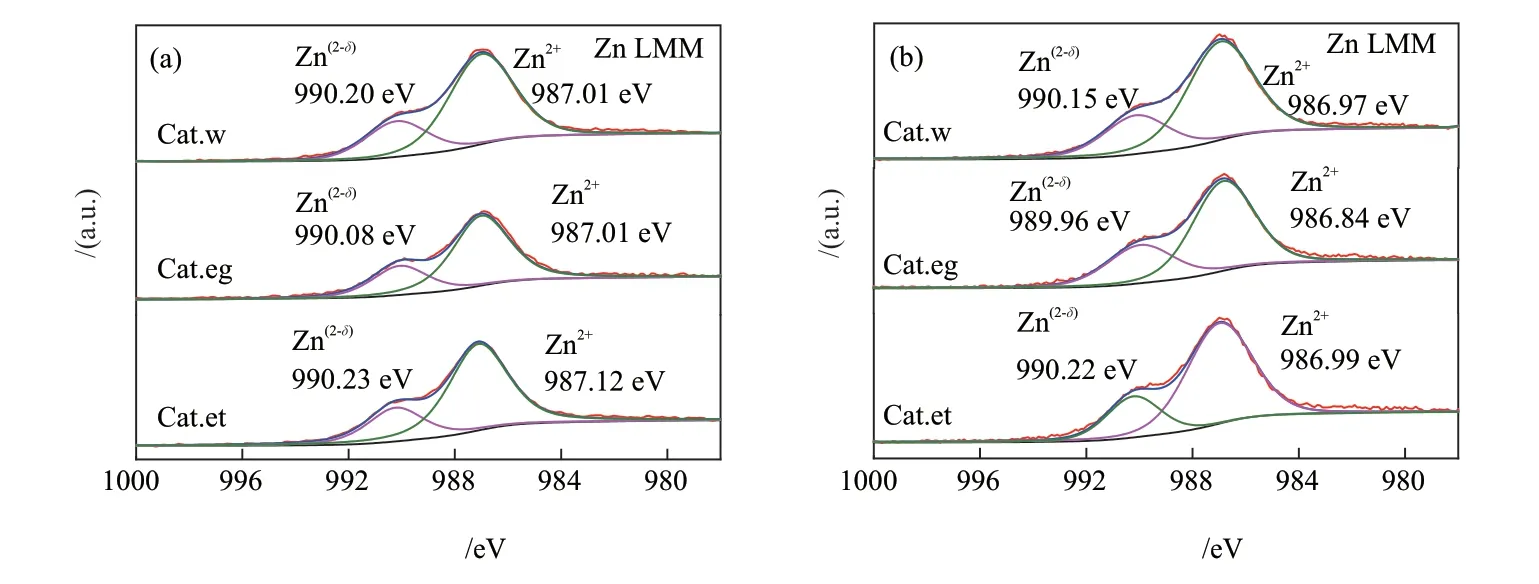
图8 反应前(a)和反应后(b)催化剂的Zn LMM俄歇谱Fig. 8 Zn LMM spectra of catalysts before (a) and after (b) reaction
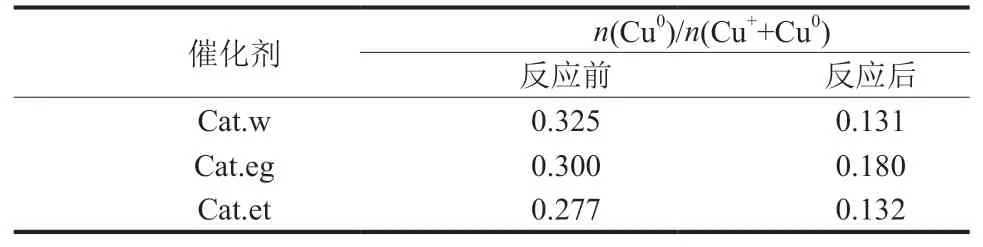
表5 俄歇谱图分峰拟合得反应前后催化剂表面不同价态Cu占比Table 5 Proportion of surface Cu with different valence in catalysts before and after reaction according to Auger spectra peaks fitting
XPS测得的催化剂反应前后表面元素含量如表7 所示。由表7 可知,表面Cu含量少于3%,C含量大于70%,这也是完全液相法的特征之一。从表面各元素原子数量占比看来,Cu元素相对量远低于其投料比(n(Cu)/n(Zn)/n(Al) = 2/1/0.8),表面Cu含量过低是CO转化率不高的主要原因。对比反应前后的表面原子数量比n(Zn)/n(Cu)、n(Al)/n(Cu)和n(Al)/n(Zn)可以发现,在反应过程中Zn和Al进一步在表面富集,且Zn的富集程度大于Al。不同溶剂催化剂Zn、Al富集程度不同,使用有机溶剂(Cat.et和Cat.eg)时,表面Zn、Al富集程度更高。但这种富集程度的变化与乙醇占比没有对应关系,即和乙醇的选择性生成没有对应关系。综上,反应初期催化剂表面Cu含量降低,Zn、Al向表面富集是催化剂CO转化率下降的根本原因。
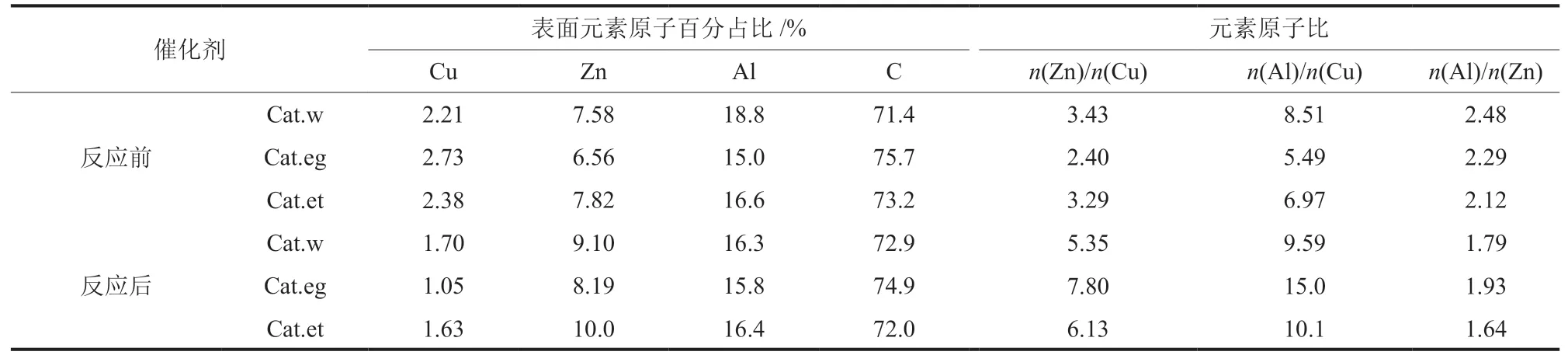
表7 反应前后催化剂表面元素原子含量Table 7 Atomic content of surface elements in catalysts before and after reaction
2.5 TEM表征
TEM表征如图9、图10 和图11 所示,可见3种催化剂在形貌上没有本质区别。以有机物做溶剂的Cat.et和Cat.eg催化剂呈不规则片状结构,反应前后形貌没有明显变化,片层中Zn、Al、O、C和Cu均匀分布。

图9 Cat.w催化剂反应前的TEM图(a)、反应后的HRTEM图(b)和TEM图(c)以及元素分布图(d)~(h)Fig. 9 TEM images (a) before reaction, HRTEM images (b) and TEM images (c) after reaction and element distribution images (d)~(h) of catalyst Cat.w

图10 Cat.eg催化剂反应前的TEM图(a)、反应后的HRTEM图(b)和TEM图(c)以及元素分布图(d)~(h)Fig. 10 TEM images (a) before reaction, HRTEM images (b) and TEM images (c) after reaction and element distribution images of(d)~(h) of catalyst Cat.eg
图10(b)和图11(b)晶格间距显示,片层中的Cu元素以Cu0的形式存在,样品中也观察到少量大颗粒Cu0存在(图10(a)、图11(a)和(d))。以水为溶剂的Cat.w形貌与上述两种催化剂有一定差别,为富含纺锤状体的片状结构,反应后其纺锤体更加明显(图9(c))。EDS分析表明,纺锤体中Cu原子百分比为8.45%,Zn为77.80%,Al为13.80%,可见其主要为ZnO构成,同时观察到有较多的大颗粒Cu物种存在,这与其投料组成对应。纺锤体中ZnO为无序态固体(图9(c)),稳定性低。文献[46]报道无序态ZnO由于界面梯度效应,能够在远离界面约2 nm处进行再次成核,随后附着在界面上生成介晶。文献[47]认为,ZnO逐渐向Cu晶粒表面上迁移,并在载体界面上从无序态变为晶态。结合本文XRD与XPS表征结果,Cat.w催化剂在反应过程中甲醇占总醇选择性不断上升,主要是由于无序ZnO迁移至Cu0表面形成介晶ZnO,覆盖在Cu0表面减小了其表面能[46-48],最终体系稳定于能量较低的状态,形成Cu-ZnO甲醇合成位点。

图11 Cat.et催化剂反应前的TEM图(a)、反应后的HRTEM图(b)和TEM图(c)和元素分布图(d)~(h)Fig. 11 TEM images (a) before reaction, HRTEM images (b) and TEM images (c) after reaction and element distribution images of(d)~(h) of catalyst Cat.et
2.6 N2 吸/脱附表征
图12为反应前后催化剂的N2吸/脱附曲线和孔径分布。由图12 可知,各催化剂N2吸附均为IV型等温线、H3 型回滞环,属于平板狭缝结构孔道,是典型的AlOOH孔结构[49],平均孔径在25 nm左右,孔径分布中4 nm的最可几孔为空化效应造成的假孔[50-52]。催化剂织构性质如表7。由表7 可知,3 种催化剂在反应后比表面积均有所增大,平均孔径略有减小。结合催化剂性能评价结果,反应后孔径较大的Cat.et和Cat.w都具有较高的乙醇占比,表明大孔径有利于乙醇生成,而大的比表面积有利于CO转化率的提高。

图12 反应前后催化剂的N2 吸/脱附等温曲线(a)和孔径分布(b)Fig. 12 N2 adsorption/desorption isotherms (a) and pore size distribution curves (b) of catalysts before and after reaction
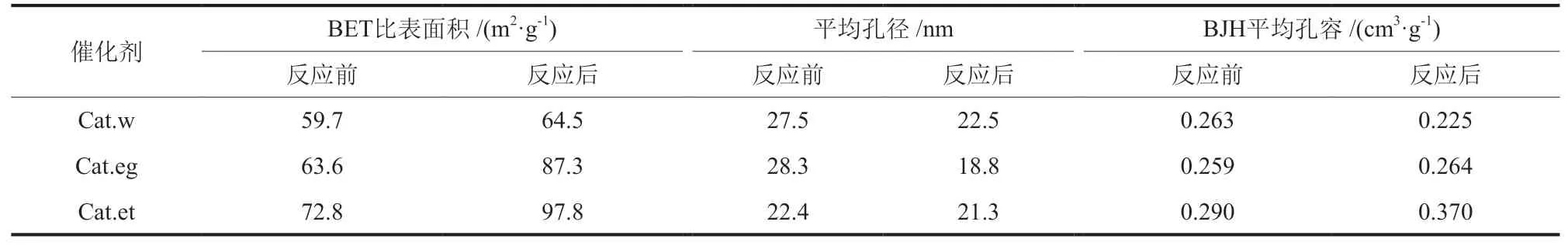
表7 反应前后催化剂的织构性质Table 7 Texture parameters of catalysts before and after reaction
2.7 O2 程序升温氧化-质谱表征
催化剂反应前后O2程序升温氧化-质谱(O2-TPO-MS)表征结果如图13,对归属于CO2质荷比44 的碎片进行了跟踪检测。由图13 可知,3 种催化剂反应前后均于350~400 °C之间存在碳的氧化峰,反应后氧化峰面积有所减少,这与前文表征结果一致,也表明催化剂表面碳物种不是导致催化剂反应初期活性降低的原因。
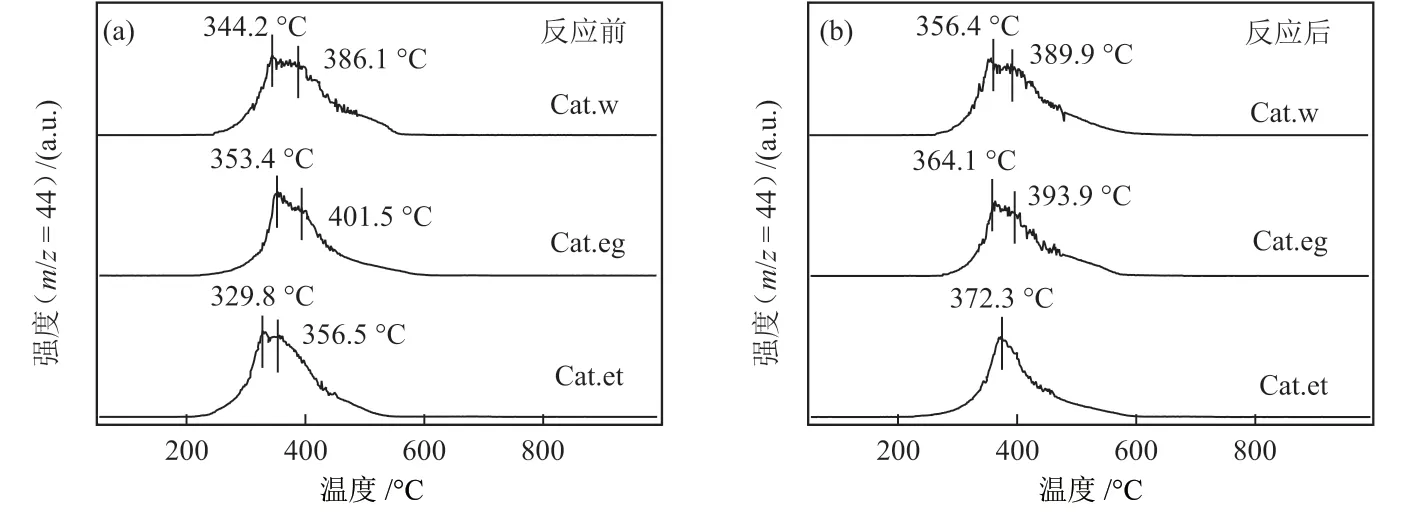
图13 反应前(a)和反应后(b)催化剂的O2-TPO-MS谱图Fig. 13 O2-TPO-MS spectra of catalysts before (a) and after (b) reaction
3 结论
在完全液相两步水解法基础上,通过改变CuZn溶胶溶剂制备了CuZnAl浆状催化剂,研究了其性能与结构和表面态的关系,得到如下主要结论。
以乙醇和乙二醇为溶剂的Cat.et和Cat.eg催化剂,反应前后催化剂均为片层状结构,结构稳定;Cat.et催化剂比表面积大,CO转化率最高,乙醇占比最高,达55.5%;Cat.eg比表面积次之,但乙醇占比最低。以水为溶剂的Cat.w催化剂,结构中存在较多以ZnO为主要成分的纺锤体,组分分布不均匀,ZnO覆盖于Cu0上,构成Cu-ZnO甲醇活性位点,比表面积最低,CO转化率最低,乙醇占比次之。孔径尺寸与乙醇占比有正相关性。
所制CuZnAl浆状催化剂上存在两种碳链增长路径,通过表面CH3物种的CO插入以及通过表面CHO的偶联,以第一种路径为主。催化剂表面高浓度Cu+是选择性生成乙醇的关键。
