高镍正极材料中钴元素的替代方案及其合成工艺优化
2022-06-21吴锋李晴陈来王紫润陈刚包丽颖卢赟陈实苏岳锋
吴锋,李晴,陈来,王紫润,陈刚,2,包丽颖,卢赟,2,陈实,苏岳锋
1北京理工大学材料学院,环境科学与工程北京市重点实验室,北京 100081
2北京理工大学重庆创新中心,重庆 401120
3劳伦斯伯克利国家实验室能量存储与分布式资源部门,美国 加州 94720
1 引言
目前,自然能源消耗日益加剧,由此导致的环境污染问题也日益严重。因此,清洁二次能源材料的发展是当前世界范围内备受瞩目的热点1-3。近年来,绿色二次电池技术的发展突飞猛进,其中具有高能量密度的锂离子电池作为储能装置已广泛应用于现代生活,在储能电网,电动工具,便捷式移动设备,以及电动汽车,电动飞机,电动轮船等均有其身影4-7。中国对锂离子电池的发展极为重视并做出了重要规划。其中《中国制造2025》中对动力电池发展规划的要求是到2020年,电池能量密度达到300 Wh·kg-1,到2025年达到400 Wh·kg-1,到2030年达到500 Wh·kg-1;《汽车产业中长期发展规划》中则制定目标为到2020年,动力电池单体能量密度达到300 Wh·kg-1,力争实现350 Wh·kg-1,系统比能量密度力争达到260 Wh·Kg-1,成本降至1元·Wh-1以下,到2025年,动力电池系统比能量达到350 Wh·kg-1。
想锂离子电池是最具潜力的候选电池体系之一要实现上述目标,以高镍层状材料为正极的8-10。高镍层状正极材料具有比能量密度高(可以达到800 Wh·kg-1),快充性能良好,环境友好等优点11-13。而高镍正极材料也存在一些缺点,比如在充放电过程中,由于Ni2+离子半径(r= 0.069 nm)和Li+离子半径(r= 0.076 nm)相近,很容易导致镍离子向锂位迁移,造成锂镍混排(即“阳离子混排”),从而破坏高镍正极材料结构并诱发电化学性能衰减14-16。
在高镍层状正极材料LiNixMnyCo1-x-yO2(x>0.8)中,不同过渡金属元素起到的作用不同。其中,Ni在电化学充放电过程中存在Ni2+↔ Ni3+↔Ni4+的氧化还原过程,这个过程伴随着更多的Li+可逆嵌入脱出晶格,为高镍正极材料提供了更高的可逆循环容量17,18;Mn4+离子在电化学过程中不发生氧化还原反应,有助于提高正极材料在电化学过程中的结构稳定性和热稳定性能19,20;而Co3+磁矩为0,占据高镍层状结构中过渡金属位置后能够抑制锂镍混排,并增强材料导离子能力21,22。
钴在高镍正极材料中的作用至关重要,但是目前全球范围内钴矿资源紧缺,存储量仅为687.5万吨,氧化钴价格更是高达21万元每吨;而原材料成本价格偏高是阻碍高镍正极材料广泛应用的主要原因之一。因此,寻找一种资源存储量高、价格低廉且不影响高镍正极材料的电化学性能的过渡金属元素替代Co,是目前商业界以及学术界共同关注的热点问题。由此,本研究根据Co的性质,选取Cr3+、Cd2+、Zr4+几种元素替代Co3+,进行无钴化高镍层状材料的相关研究,分析其可行性并进行合成工艺优化,最后对通过合成更高镍含量的无钴正极材料验证该优化工艺。本实验的设计思路如图1所示。本文旨在通过共沉淀法合成高性能的无钴化高镍正极材料,以期降低锂离子电池正极材料的成本,从而促进高镍材料在电动汽车中的广泛应用。
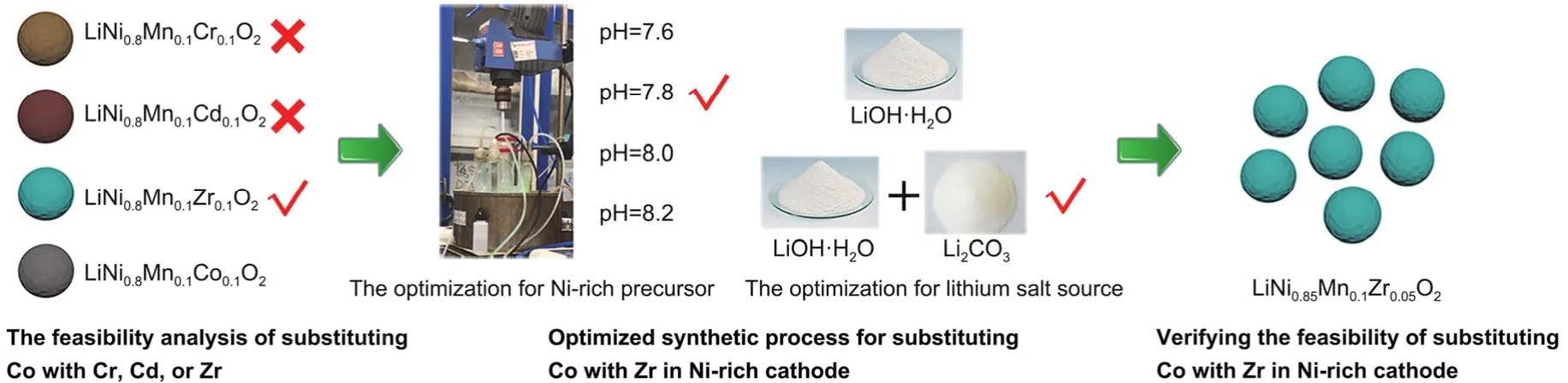
图1 实验设计思路Fig. 1 The design idea of the experiment.
2 实验部分
2.1 样品制备
样品的具体合成条件如表1所列。
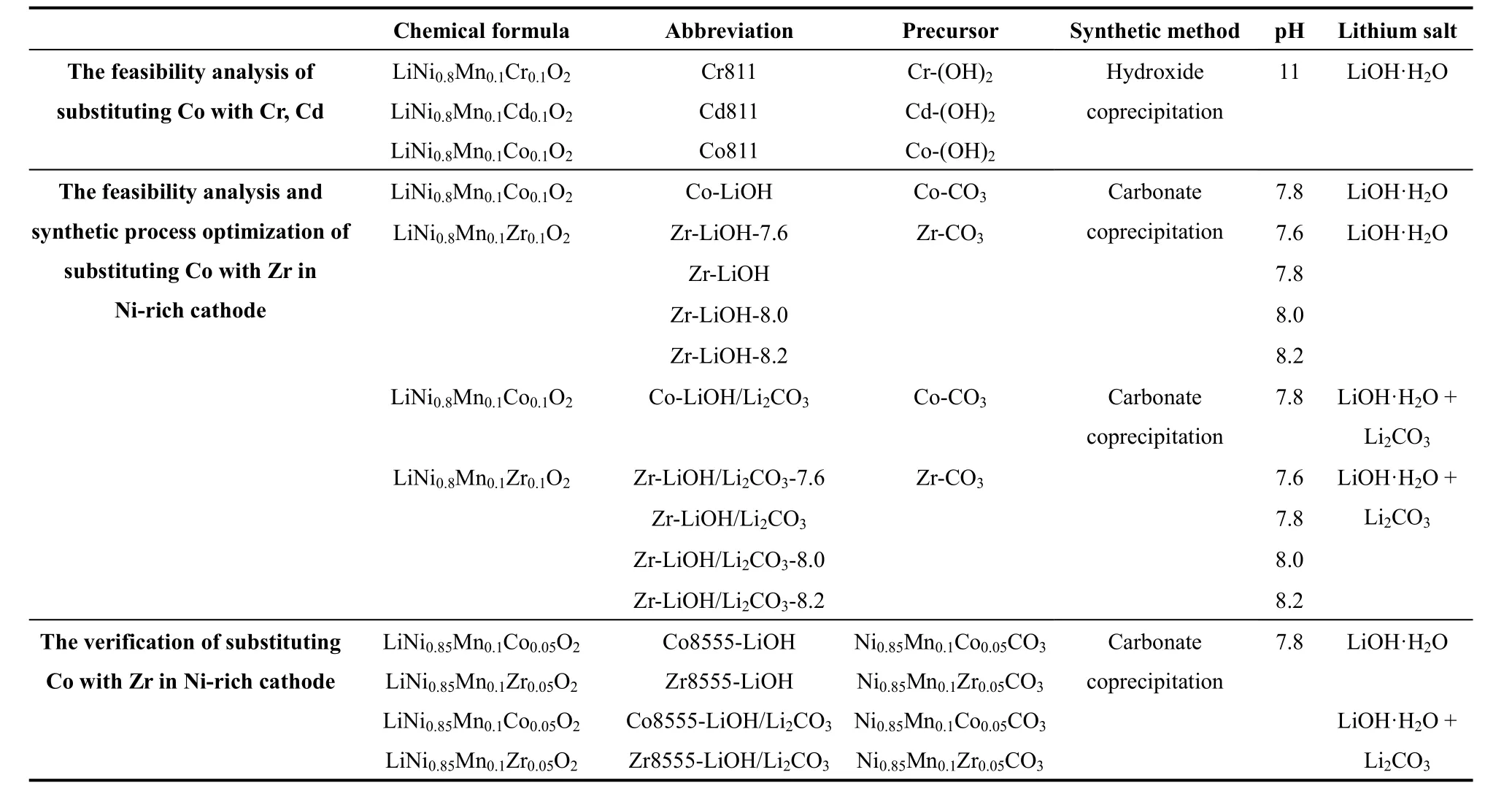
表1 样品合成条件列表Table 1 Synthesis conditions for all samples.
通过氢氧化物共沉淀法,制备Ni0.8Mn0.1M’0.1(OH)2(M’ = Cr,Cd,Co)前 驱体。实验步骤如文献所述16。通过氢氧化物共沉淀法得到的前驱体Ni0.8Mn0.1Cr0.1(OH)2,Ni0.8Mn0.1Cd0.1(OH)2和Ni0.8Mn0.1Co0.1(OH)2分 别 记为Cr-(OH)2,Cd-(OH)2和Co-(OH)2。
通过碳酸盐共沉淀方法合成Ni0.8Mn0.1M”0.1CO3(M” = Zr,Co)前驱体。按照化学计量比将Ni,Mn和M” (M = Co,Zr)的硫酸盐溶于去离子水并配置成2 mol·L-1溶液A,再配置一定浓度的无水碳酸钠溶液B,分别将A和B两种溶液搅拌0.5 h后同时泵入55 °C水浴的圆底烧瓶中,通入适量氨水调节pH至7.8。进样结束后,持续搅拌2 h使前驱体颗粒充分生长,经去离子水洗涤、过滤,将得到的滤渣在真空烘箱中干燥12 h,得到碳酸盐前驱体,分别记为Zr-CO3和Co-CO3。
本研究中采用两种前驱体混入锂化方法。方法一采用LiOH·H2O作为锂源,按照nLi:nTM=1.02 : 1 (TM为过渡金属元素)的比例将锂盐与前驱体研磨均匀后,以550 °C初烧5 h,之后以730 °C复烧12 h,得到高镍正极材料。其中,以氢氧化物前驱体为原料制得的高镍正极材料记为Cr811、Cd811、Co811,以碳酸盐前驱体为原料制得的高镍正极材料分别记为Zr-LiOH和Co-LiOH。方法二同时使用LiOH·H2O和Li2CO3为锂源,按照nLi:nTM= 1.02 : 1,nLiOH·H2O:nLi2CO3= 1 : 1的比例将锂盐与碳酸盐前驱体混合研磨均匀,以550 °C初烧5 h,之后以750 °C复烧12 h,得到高镍正极材料,分别记为Zr-LiOH/Li2CO3和Co-LiOH/Li2CO3。
进一步的,在上述碳酸盐共沉淀方法合成Ni0.8Mn0.1Zr0.1CO3前驱体过程中,为进一步探究最佳反应pH值,分别用氨水调节pH为7.6,7.8,8.0和8.2。而后采用方法一和方法二进行锂化烧结,其中混入LiOH·H2O (nLi:nTM= 1.02 : 1),经过研磨和高温煅烧(550 °C初烧5 h,730 °C复烧12 h)得到正极材料,分别记为Zr-LiOH-7.6,Zr-LiOH,Zr-LiOH-8.0和Zr-LiOH-8.2;混入LiOH·H2O和Li2CO3的混合盐(nLi:nTM= 1.02 : 1,nLiOH·H2O:nLi2CO3= 1 : 1),经过研磨和高温煅烧(550 °C初烧5 h,750 °C复烧12 h)得到的正极材料分别记为Zr-LiOH/Li2CO3-7.6,Zr-LiOH/Li2CO3,Zr-LiOH/Li2CO3-8.0和Zr-LiOH/Li2CO3-8.2。
在上述碳酸盐共沉淀方法中,通过氨水调节pH为7.8,调整过渡金属离子摩尔比例为Ni : Mn :M” = 0.85 : 0.1 : 0.05,合成前驱体Ni0.85Mn0.1Co0.05CO3和Ni0.85Mn0.1Zr0.05CO3。 混 入锂盐LiOH·H2O (nLi:nTM= 1.02 : 1),经过研磨和高温煅烧(550 °C初烧5 h,730 °C复烧12 h)得到的正极材料记为Co8555-LiOH,和Zr8555-LiOH。混入锂盐LiOH·H2O和Li2CO3的混合盐(nLi:nTM=1.02 : 1,nLiOH·H2O:nLi2CO3=1 : 1),经过研磨和煅烧(550 °C初烧5 h,750 °C复烧12 h)得到的正极材料记为Co8555-LiOH/Li2CO3和Zr8555-LiOH/Li2CO3。
原料 :MnSO4·H2O (分析纯,99%),CoSO4·7H2O (分析纯,99%),NiSO4·7H2O (分析纯 ,99%),Zr2(SO4)2·4H2O (分析纯,98%),Cr2(SO4)3·xH2O (分析纯,99%),CdSO4·8/3H2O(分析纯,99%)。
2.2 样品表征
采用旋转阳极X射线衍射仪(XRD,日本Rigaku公司,型号IV-185)分析样品材料结构,测试条件:Cu靶,Kα射线,靶电压40 kV,靶电流40 mA,扫描范围为10°-90°,2θ扫描速率为8 (°)·min-1。 采用荷兰公司生产的扫描电镜(SEM,型号QUANTA 250),分析目标产物的颗粒大小和表面形貌。采用电感偶合等离子体原子发射光谱(ICP-AES,IRIS Intrepid II),测量层状氧化物中各元素的成分比例。
2.3 电池组装和测试
将正极活性物质和聚偏氟乙烯(PVDF)以及乙炔黑按照质量比8 : 1 : 1混合成浆料,以N-甲基-2-吡咯烷酮(NMP)为粘结剂,研磨均匀后,涂布在铝集流体上,置于80 °C烘箱中干燥10 h,经过辊压裁片,在手套箱中组装成CR2025型扣式电池。其中负极为金属锂片,隔膜为Celgard2300多孔复合聚合物膜,电解液为1 mol·L-1的LiPF6溶解于碳酸乙烯酯(EC)和碳酸二甲酯(DMC) (VEC:VDMC= 1 :1)。电池测试在LAND电池测试系统(武汉金诺公司)上于25 °C进行恒电流充放电循环测试,充放电电压区间为2.75-4.3 V (vs.Li/Li+),其中充放电电流以1C=180 mA·g-1计算。
3 结果与讨论
3.1 Cr3+和Cd2+掺杂的无钴高镍正极材料
由于Cr3+的离子半径(r= 0.0615 nm)与Co3+离子半径(r= 0.0545 nm)相近,Cr与Co在元素周期表中位于同一主族,且Cr3+掺杂可以有效增强高镍层状正极材料电化学性能23;另外,由于电荷补偿作用,Cd2+掺杂进入层状正极晶格可以提高Ni离子的氧化价态,从而减少Li+/Ni2+混排,因此选择Cr3+和Cd2+作为替代Co的元素。
Cr-(OH)2、Cd-(OH)2和Co-(OH)2三种前驱体材料的形貌如图2中SEM图所示,Cr-(OH)2氢氧化物前驱体二次颗粒直径4 μm,且孔洞比较多,不利于电化学循环过程中Li+的可逆嵌脱;Cd-(OH)2氢氧化物前驱体二次颗粒直径为8 μm,二次颗粒尺寸均匀但一次颗粒呈现比较大的薄片状结构,且一次颗粒的排布无序同样不利Li+的嵌脱;Co-(OH)2的氢氧化物前驱体二次颗粒直径10 μm,二次颗粒尺寸分布均匀且一次颗粒呈放射状向心排布,能够致密堆积形成二次颗粒,有利于充放电过程中Li+的迁移。

图2 氢氧化物共沉淀前驱体Cr-(OH)2、Cd-(OH)2、Co-(OH)2的SEM图Fig. 2 SEM images of (a) Cr-(OH)2, (b) Cd-(OH)2, and(c) Co-(OH)2.
采用电感耦合等离子体原子发射光谱(ICP)定量分析Cr811,Cd811和Co811三种正极材料的元素组成,其化学式分别为LiNi0.792Mn0.081Cr0.127O2,LiNi0.777Mn0.087Cd0.136O2和LiNi0.810Mn0.093Co0.097O2,与Ni : Mn : M’ (M’ = Cr, Cd, Co) = 8 : 1 : 1的情况基本吻合。在2.75-4.3 V电压范围内测试三种正极材料的电化学循环性能,如图3所示,Co811的初始放电容量188.1 mAh·g-1,循环100周容量保持率为87.7%;而Cd811的初始放电容量是84.3 mAh·g-1,100周容量保持率57.2%;Cr811材料的初始放电容量只有65.9 mAh·g-1,100周容量保持率仅为61%。由于Cd和Cr替代Co得到的材料电化学性能都比较差,因此,我们认为Cd和Cr在高镍三元材料中替代Co是不可行的。

图3 Co811,Cd811和Cr811的0.2C电化学循环性能图Fig. 3 Electrochemical cycling performance of Co811,Cd811, and Cr811 at 0.2C.
3.2 Zr4+掺杂的无钴高镍正极材料
由于Zr4+离子掺杂高镍正极材料可以有效提高高镍正极材料结构稳定性,抑制其阳离子混排,提高正极材料导电性,又因为全球范围内锆矿(ZrO2)储量稳定在7500万吨,并且价格低廉,高级锆英砂每吨仅1万元左右,远低于钴矿价格24-26。所以Zr可以作为另一个替代Co的过渡金属元素掺杂到高镍正极材料中。由于Zr4+沉淀pH值的限制,我们采用碳酸盐共沉淀法合成碳酸盐前驱体Zr-CO3以及其对比样Co-CO3。通过SEM测试分析这两种前驱体材料形貌(图4),Zr-CO3碳酸盐共沉淀前驱体的颗粒破碎严重,成球性较差,相比较而言Co-CO3碳酸盐共沉淀前驱体材料二次颗粒成球度更高,一次颗粒堆积的更致密。

图4 碳酸盐共沉淀前驱体(a) Zr-CO3和(b) Co-CO3的SEM图Fig. 4 SEM images of (a) Zr-CO3 and (b) Co-CO3.
3.2.1 Zr4+掺杂无钴高镍正极材料的锂化工艺优化
ICP测得Co-LiOH和Co-LiOH的化学式分别为LiNi0.76Mn0.11Zr0.13O2, 和LiNi0.810Mn0.094Co0.096O2,符合预期设计。对这两种材料进行电化学性能测试,如图5和表2所示。在0.2C倍率下,Co-LiOH在2.75-4.3 V的首周放电比容量为157.5 mAh·g-1,100周容量保持率86.1%。碳酸盐共沉淀法合成的Co-LiOH的电化学性能略差于氢氧化物共沉淀法得到的Co811,因此本实验后面进一步探究了碳酸盐共沉淀合成高镍材料前驱体的优化条件。Zr-LiOH正极材料的首周放电容量为145.8 mAh·g-1,100周容量保持率为87.04%,虽然其放电比容量略低于Co-LiOH,但是其容量保持率却有所改善。

图5 Co-LiOH和Zr-LiOH的(a) 0.2C长循环测试图和(b) 倍率性能测试图;Co-LiOH/Li2CO3和Zr-LiOH/Li2CO3的(c) 0.2C长循环测试图和(d)倍率性能测试图Fig. 5 (a) Electrochemical cycling performance at 0.2C and (b) Rate performance of Co-LiOH and Zr-LiOH;Electrochemical cycling performance at 0.2C and (b) Rate performance of Co-LiOH/Li2CO3 and Zr-LiOH/Li2CO3.
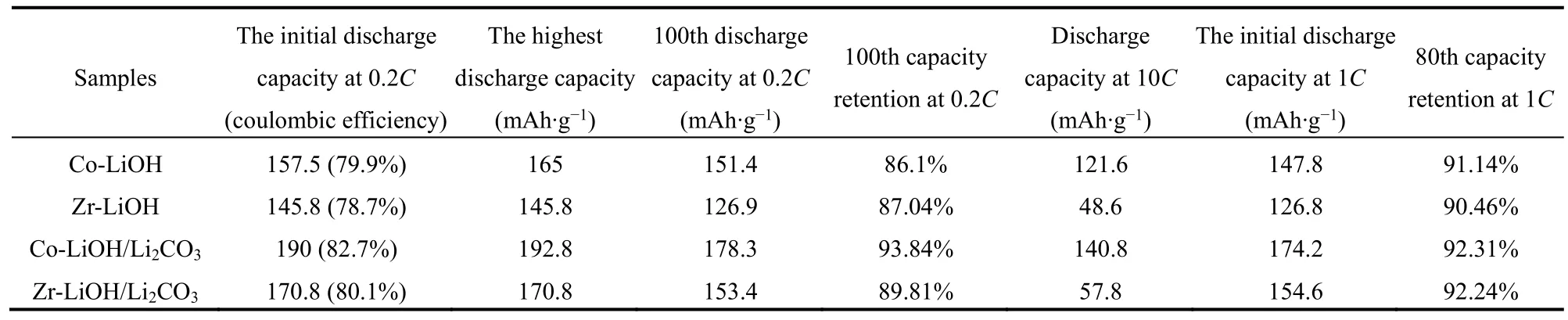
表2 Co-LiOH,Zr-LiOH,Co-LiOH/Li2CO3 和Zr-LiOH/Li2CO3的电化学性能表Table 2 Electrochemical performances for Co-LiOH, Zr-LiOH, Co-LiOH/Li2CO3 and Zr-LiOH/Li2CO3.
对Co-LiOH和Zr-LiOH进行倍率性能测试(在0.2C、0.5C、1C、2C、5C和10C分别循环3周,然后恢复至0.2C循环3周,再在1C循环80周),结果如图5b所示 。Co-LiOH在0.2C、0.5C、1C、2C、5C和10C的放电容量分别为160、156.2、149.9、144.7、133.5、121.6 mAh·g-1,并在恢复0.2C循环时表现出160.9 mAh·g-1的放电容量。之后的80周1C循环中,Co-LiOH的初始放电容量和80周容量保持率分别为147.8 mAh g-1和91.14%。相比之下,Zr-LiOH的倍率性能略显逊色,在倍率充放电0.2C、0.5C、1C、2C、5C和10C分 别 为146.5、132.8、118.2、104.9、73.9、48.6 mAh·g-1,并在恢复0.2C循环时表现出141 mAh·g-1的放电容量。在接下来的1C循环中,Zr-LiOH初始放电容量和80周容量保持率分别为126.8 mAh·g-1和90.46%。可以看到,Zr替代Co合成的811型高镍材料,虽然放电容量仍然不足以与NCM811材料媲美,但其循环稳定性比较好,因此仍有进一步探究Zr替代Co掺杂进入高镍正极材料可行性的必要性。
不同锂源的选择会影响前驱体材料转化成层状正极材料的结晶过程,从而影响成品材料的电化学性能。以LiOH·H2O为锂盐时,前驱体材料锂化结晶转变成层状正极材料所需要的温度相比于以Li2CO3为锂盐的情况所需的温度低,因而一般以LiOH·H2O为锂源合成三元正极材料所需的煅烧温度低于以Li2CO3为锂源所需的煅烧温度。在加热过程中LiOH·H2O会发生分解形成Li2O,导致以LiOH·H2O为锂盐形成的高镍正极材料二次颗粒表面残锂相多于以Li2CO3为锂盐合成的高镍正极材料二次颗粒的表面残锂相27。为了进一步优化无钴高镍正极材料的性能,我们选择不同锂盐为锂源合成高镍正极材料,并分析其电化学性能。
Zr-LiOH/Li2CO3和Co-LiOH/Li2CO3在0.2C,2.75-4.3 V电压范围内的电化学性能如图5c和表2所示。Co-LiOH/Li2CO3在2.75-4.3 V的初始放电容量190 mAh·g-1,100周容量保持率93.84%。而Zr-LiOH/Li2CO3正极材料的首周放电容量170.8 mAh·g-1,100周容量保持率为89.81%。两种材料在不同倍率下的充放电性能如图5d所示,在电压2.75-4.3 V范围内,Co-LiOH/Li2CO3在0.2C、0.5C、1C、2C、5C和10C倍率下的放电容量分别为184、181.2、173.7、165.7、152.6和140.8 mAh·g-1, 并在恢复0.2C循环时表现出188 mAh·g-1的放电容量。相比之下,Zr-LiOH/Li2CO3的倍率性能略差,在倍率0.2C、0.5C、1C、2C、5C和10C下的放电容量分别为183、155.2、141、110.7、81.4和57.8 mAh·g-1,并在恢复0.2C循环时表现出175.5 mAh·g-1的放电容量。
在接下来的1C循环中,Co-LiOH/Li2CO3和Zr-LiOH/Li2CO3都展现了较好的容量保持率,Co-LiOH/Li2CO3的初始1C放电容量和80周容量保持率 分 别 为174.2 mAh·g-1和92.31%;Zr-LiOH/Li2CO3的初始1C放电容量和80周容量保持率分别154.6 mAh·g-1和92.24%。 可 以 看 到Zr-LiOH/Li2CO3的电化学循环稳定性能优于Co-LiOH/Li2CO3。由于以LiOH·H2O和Li2CO3混合盐作为锂源得到高镍正极材料的锂化结晶程度好,二次颗粒表面残锂相相对较少,所以Zr-LiOH/Li2CO3的电化学性能优于Zr-LiOH。
3.2.2 Zr4+掺杂无钴高镍正极合成过程pH优化
在碳酸盐共沉淀合成前驱体过程中,pH值对材料形貌以及性能影响很大,pH值过低会导致在共沉淀体系中过渡金属离子络合平衡占据主导,晶体生长速度快,成核相对缓慢,从而导致颗粒形貌过大;而若pH值过高,则会使得体系中碳酸盐沉淀过饱和度增大,成核速度快,晶核来不及长大,从而得到的材料颗粒尺寸较小28-30。因此应进一步探究Zr掺杂的无钴高镍正极材料合成的最优pH条件。通过调节氨水用量控制碳酸盐共沉淀合成过程中的pH分别为7.6、7.8、8.0和8.2,并探究此种情况下合成的材料在2.75-4.3 V,0.2C测试条件下测得的电化学性能,如图6所示。
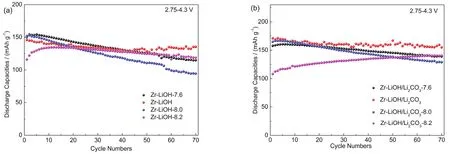
图6 (a) Zr-LiOH-7.6,Zr-LiOH,Zr-LiOH-8.0和 Zr-LiOH-8.2;(b)Zr-LiOH/Li2CO3-7.6,Zr-LiOH/Li2CO3,Zr-LiOH/Li2CO3-8.0和Zr-LiOH/Li2CO3-8.2在0.2C的电化学循环性能图Fig. 6 Electrochemical cycling performance of (a) Zr-LiOH-7.6, Zr-LiOH, Zr-LiOH-8.0, and Zr-LiOH-8.2;(b) Zr-LiOH/Li2CO3-7.6, Zr-LiOH/Li2CO3, Zr-LiOH/Li2CO3-8.0, and Zr-LiOH/Li2CO3-8.2 at 0.2C.
如图6a和表3所示, Zr-LiOH-7.6的初始放电比容量为151.6 mAh·g-1,经过活化后最高放电比容量为154.3 mAh·g-1(第5周),循环70周后容量保持率为74.4%;Zr-LiOH的初始放电容量145.8 mAh·g-1(首周为最高放电容量),循环70周容量保持率为92.73%;Zr-LiOH-8.0的初始放电容量149.9 mAh·g-1,最高放电容量为152.8 mAh·g-1(第2周),循环70周容量保持率为61.71%;Zr-LiOH-8.2的初始放电容量116 mAh g-1,最高放电容量为134.6 mAh·g-1(第12周),循环70周容量保持率为88.26%。由此可知,Zr-LiOH-7.6的初始放电容量最高,但循环稳定性较差;Zr-LiOH-8.0的放电容量和循环性能均较为恶劣;虽然Zr-LiOH-8.2的容量保持率高,但是其放电容量整体偏低;而Zr-LiOH的放电容量和循环容量保持率都比较理想。
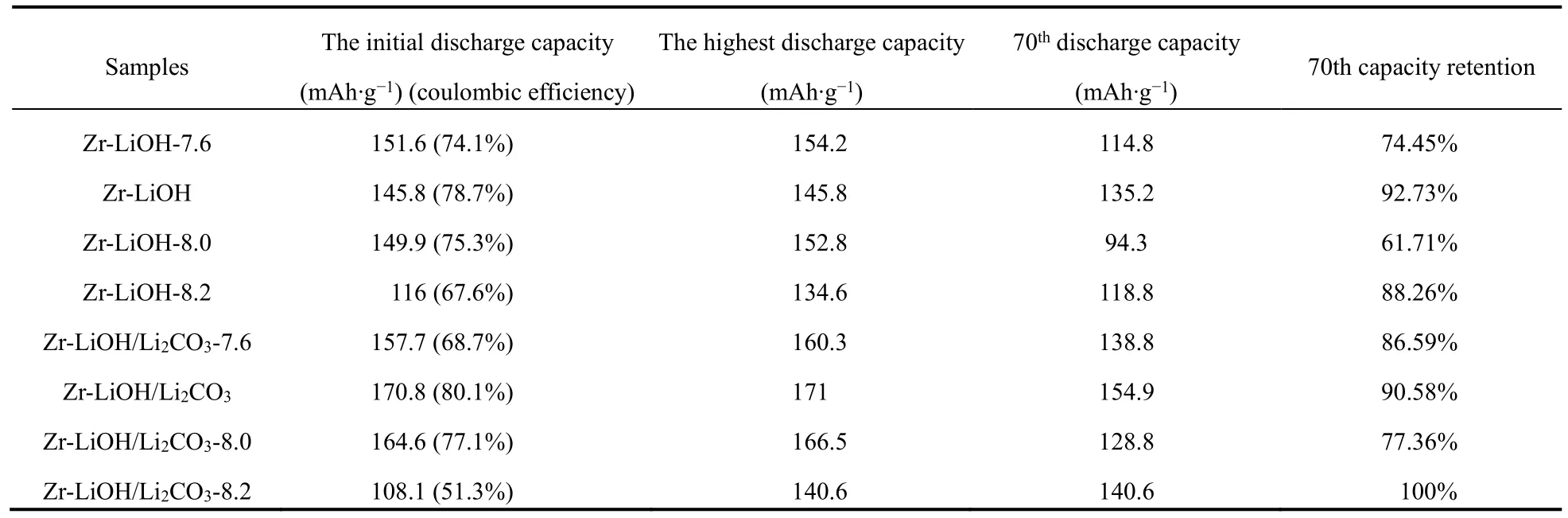
表3 在不同pH值条件下合成的Zr4+掺杂的无钴高镍正极材料的电化学性能Table 3 Electrochemical performances of Zr4+ doped Cobalt-free Ni-rich cathode materials synthesized at different pH.
使用LiOH·H2O和Li2CO3混合锂源锂化得到的高镍正极材料的电化学性能如图6b和表3所示。Zr-LiOH/Li2CO3-7.6的初始放电容量157.7 mAh·g-1,最高放电容量为160.3 mAh·g-1(第5周), 循环70周容量保持率为86.59%;Zr-LiOH/Li2CO3的初始放电容量170.8 mAh·g-1,最高放电容量为171 mAh·g-1(第3周),循环70周容量保持率为93.16%;Zr-LiOH/Li2CO3-8.0的初始放电容量164.6 mAh·g-1,最高放电容量为166.5 mAh·g-1(第4周),循环70周容量保持率为77.36%;Zr-LiOH/Li2CO3-8.2的初始放电容量108.1 mAh·g-1,最高放电容量为140.6 mAh·g-1(第70周)。由此可知,Zr-LiOH/Li2CO3正极材料的放电容量和循环稳定性能都是最佳的。相对比与LiNi0.8Co0.1Mn0.1O2正极材料,Zr掺杂形成的无钴正极材料放电容量偏低,但电化学循环性能良好。
通过电化学性能的测试分析,可以认定7.8是碳酸盐共沉淀合成Zr掺杂高镍正极前驱体材料的最适宜pH值,此时碳酸盐沉淀的成核和晶体生长速度达到平衡,得到的材料中各过渡金属元素可以均匀沉淀。因而在采用不同锂源的高镍正极材料中,在pH = 7.8合成的Zr-LiOH和Zr-LiOH/Li2CO3电化学性能都是最优秀的。另外,通过比较不同pH值合成的前驱体材料分别以LiOH·H2O和LiOH·H2O和Li2CO3混合盐作为锂源,经过高温煅烧得到的正极材料的电化学性能发现,所有以混合盐作为锂源得到的高镍正极材料的电化学性能都优于单独使用LiOH·H2O作为锂盐的正极材料。这是因为以混合锂盐作为锂盐的无钴高镍正极材料的锂化结晶程度最好,表面残锂相最少,所以经过合成工艺优化得到的Zr-LiOH/Li2CO3的电化学性能最优。
3.2.3 合成更高镍含量的无钴正极材料验证
通过合成更高镍含量的无钴高镍正极材料LiNi0.85Mn0.1Zr0.05O2来验证优化合成工艺的可行性。镍含量更高的正极材料具有更高的放电比容量,但是高镍正极材料的结构稳定性却随着镍含量的增加而降低。首先通过XRD分析Co-LiOH,Zr8555-LiOH,Co8555-LiOH/Li2CO3和Zr8555-LiOH/Li2CO3四种材料的晶体结构。如图7所示,这四种材料的XRD主峰位置与图7下方竖线对应的层状结构LiNiO2(PDF#09-063)吻合,说明通过碳酸盐共沉淀(pH = 7.8)合成前驱体再经过锂化煅烧可以得到空间群为的层状正极材料,且四种材料的(108)和(110)峰,以及(006)和(102)峰之间的分峰清晰明显,表明其层状晶格结构良好31。四种材料的XRD图谱都没有杂峰存在,说明合成过程中过渡金属离子均掺杂进入层状结构内部,而没有形成其他杂相。
图7 Co8555-LiOH,Zr8555-LiOH,Co8555-LiOH/Li2CO3和Zr8555-LiOH/Li2CO3的XRD测试图谱Fig. 7 The XRD patterns of Co8555-LiOH, Zr8555-LiOH,Co8555-LiOH/Li2CO3 and Zr8555-LiOH/Li2CO3.
进一步通过SEM和元素面扫分析Zr8555-LiOH/Li2CO3的形貌以及过渡金属元素分布情况,如图8所示。可以看到Zr8555-LiOH/Li2CO3正极材料二次颗粒形貌为直径10 μm的球形颗粒。其过渡金属元素Ni、Mn和Zr均匀的分布在二次颗粒表面,这也表明了Ni,Mn和Zr三种过渡金属元素在碳酸盐共沉淀过程中可以均匀的沉淀在高镍正极前驱体材料中,再经由锂化煅烧均匀的分布在高镍正极材料二次颗粒中。

图8 Zr8555-LiOH/Li2CO3的SEM以及Ni,Mn,Zr的面扫EDS图Fig. 8 The SEM image and Ni, Mn, Zr elemental mapping of Zr8555-LiOH/Li2CO3.
分别在2.75-4.3 V电压范围内,以0.2C测试不同锂化方式得到的Co8555-LiOH、Zr8555-LiOH、Co8555-LiOH/Li2CO3和Zr8555-LiOH/Li2CO3四 种材料的电化学性能,如图9a所示。Co8555-LiOH的初始放电容量163.9 mAh·g-1,最高放电容量为169.9 mAh·g-1(第2周),循环30周容量保持率为77.40%;Zr8555-LiOH的初始放电容量159.7 mAh·g-1,循环30周容量保持率为79.84%。如图9b所示,Co8555-LiOH/Li2CO3的初始放电容量181.1 mAh·g-1,循环80周容量保持率为92.44%;Zr8555-LiOH/Li2CO3的初始放电容量179.9 mAh·g-1,最 高放电容量为181.2 mAh·g-1(第5周),循环80周容量保持率为96.52%。由此可见,对于Ni : Mn : Zr/Co = 0.85 : 0.1 : 0.05的高镍正极材料,以LiOH·H2O和Li2CO3混合盐为锂源合成的材料电化学性能仍优于以LiOH·H2O为锂源合成的正极材料。
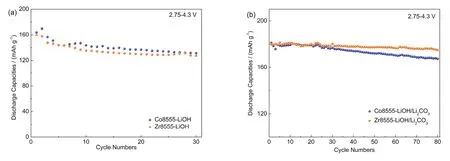
图9 (a) Co8555-LiOH和Zr8555-LiOH;(b) Co8555-LiOH/Li2CO3和Zr8555-LiOH/Li2CO3在0.2C的电化学循环性能图Fig. 9 Electrochemical cycling performance of (a) Co8555-LiOH and Zr8555-LiOH; (b) Co8555-LiOH/Li2CO3 and Zr8555-LiOH/Li2CO3 at 0.2C.
通过优化工艺合成Zr替代Co的高镍正极材料LiNi0.85Mn0.1Zr0.05O2具有优异的电化学循环稳定性,且其初始放电比容量与镍钴锰高镍三元正极材料放电容量相当。
4 结论
本文首先采用Cr,Cd和Zr替代Co合成无钴高镍正极材料,实验发现Cr和Cd替代Co的方案不可行,而Zr替代Co具有一定可行性。为进一步优化Zr替代Co合成的无钴高镍正极材料的电化学性能,本文进一步优化了其合成工艺。首先通过碳酸盐共沉淀法,合成含有Ni、Mn和Zr的前驱体正极材料。然后分别以LiOH·H2O和LiOH·H2O/Li2CO3的混合盐为锂源,发现以混合盐为锂源合成的正极材料锂化结晶程度高,表面残锂相少,电化学性能好。再调控碳酸盐共沉淀合成前驱体过程中的pH值,发现当pH = 7.8时Ni、Mn和Zr的碳酸盐共沉淀成核和晶体生长的速率达到平衡状态,此时合成的前驱体材料中过渡金属元素可以均匀沉淀,经过高温煅烧得到的正极材料电化学性能好。最后,通过验证发现该优化合成工艺仍然适用于合成更高镍含量的无钴正极材料。采用碳酸盐共沉淀(pH = 7.8)合成前驱体,经过混锂(以混合锂盐为锂源)煅烧,得到的无钴正极材料LiNi0.85Mn0.1Zr0.05O2层状结构好,锂化程度高,表面残锂相少,电化学性能优异。其首周放电容量为179.9 mAh·g-1,循环80周容量保持率为96.52%,其电化学性能略优于同等条件下合成的LiNi0.85Mn0.1Co0.05O2高镍正极材料。本实验证明Zr可以作为一种替代Co的元素掺杂进入高镍层状正极材料中,得到的无钴高镍正极材料LiNi0.85Mn0.1Zr0.05O2具有较好的电化学性能和较低的成本。
