胆道闭锁潜在分子亚型及其核心基因识别
2022-06-20陈治宏罗凤媛马丽媛李红东
陈治宏,罗凤媛,马丽媛,李红东
(1. 赣南医学院2019级硕士研究生;2. 赣南医学院2020级硕士研究生;3. 赣南医学院2021级硕士研究生;4. 赣南医学院信息工程学院,江西 赣州 341000)
胆道闭锁(Biliary atresia,BA)是新生儿常见的一种进行性阻塞性胆道疾病,临床表现为梗阻性黄疸,如果BA 患儿得不到及时治疗,易发展为胆汁淤积性肝硬化、门静脉高压,最终导致肝衰竭而死亡[1]。胆道闭锁具有地区和种族差异,一般认为亚洲人发病率较高,尤以日本和中国的发病率最高[2]。目前对BA 患儿应用最广泛且疗效较好的治疗手段是Kasai 手术,但由于进行性肝纤维化、胆汁淤积性肝硬化和门脉高压的出现,多数患儿最终须选择肝脏移植救治[3]。BA 因其早期诊断困难、病理改变特殊以及治疗效果不理想,已成为威胁新生儿生命健康的重要危险因素。
目前,研究者提出了许多理论来解释BA的病因,包括遗传变异、病毒感染和免疫介导等。CHENG G等[4]对89 例BA 患者开展全基因组拷贝数突变分析发现了29 个BA 相关的拷贝数变异,其中包括富集在炎症相关通路的基因,提示了炎症在BA 中的重要作用。SHEN C 等[5]研究表明,60%的BA 患儿在诊断时检测到巨细胞病毒(CMV)的DNA,同时感染CMV 的BA 患儿患有胆管炎和较高的胆管纤维化程度。SQUIRES J E 等[6]通过对轮状病毒(RV)诱导的BA小鼠模型研究发现,BA小鼠的NK细胞数量增加会促进慢性胆管炎症。SAXENA V 等[7]发现RV 诱导的BA 小鼠模型中的浆细胞样树突状细胞(pDCs)显著增加,同时pDCs产生白细胞介素15(IL-15)激活CD80,加速胆管纤维化进程。BA 的复杂致病因素,提示BA中可能存在不同的分子亚型。
目前,基于高通量数据对疾病进行分子亚型的研究主要是根据样本基因表达水平的相似性。通过无监督聚类的方法,例如非负矩阵分解(NMF)、层次聚类和潜在因子分析(LF)等挖掘潜在的分子亚型[8-9]。但这类方法往往基于较为复杂的数量统计模型,难以理解和进行生物学解释,且不易进行临床实践和验证。有研究表明,基因间相对表达秩次(大小)关系在正常组织样本内存在广泛的稳定性,在疾病组织样本中受到了广泛的扰动。基于这一现象,本文提出了一种基于样本内基因间相对表达秩次关系的方法识别潜在疾病分子的亚型方法。这种方法不仅易于理解和解释,还可整合来自不同实验室的样本进行生物信息学分析,对基因表达水平的个体间生物变异具有鲁棒性[10-11]。本研究将该方法应用于挖掘BA 潜在的分子亚型,进而识别亚型相关的核心基因。
1 材料与方法
1.1 数据来源及其预处理本文所分析的基因表达谱数据集,GSE46960 和GSE15235 均来自基因表达综合数据库(GEO,https://www.ncbi.nlm.nih.gov/geo/)。GSE46960数据集检测平台为GPL6244,共检测了64 例BA 患儿的肝组织、7例正常婴儿的肝组织。GSE15235 数据集检测平台为GPL570,共检测了43例BA患儿的肝组织。
各数据集均从GEO 下载原始表达谱数据(. CEL 文件),利用R 软件Affy 软件包提供的RMA(Robust Multichip Analysis)算法对其进行背景校正[12],并利用相应的平台注释文件将探针ID 对应到基因ID 上。如果多个探针映射到同一基因,则取探针的平均表达值作为该基因的表达水平。最终,数据集GSE46960 共检测18 859 个基因,数据集GSE15235 共检测20 684 个基因。两套数据集共同检测的17 179个基因作为背景基因用作后续分析。
1.2 基于基因间相对表达秩次关系识别BA 分子亚型假设样本内基因间相对表达秩次关系与疾病的分子亚型相关,那么应存在以下几种现象:⑴在不同分子亚型样本内,存在基因间相对表达秩次关系不同的现象。⑵具有潜在鉴别疾病分子亚型能力的基因对,按其相对表达秩次关系划分的两组样本间存在差异表达的基因,同时涉及生物学功能的改变。⑶识别具有鉴别疾病分子亚型能力的潜在基因对存在聚集现象。
基于上述假设,本文识别疾病分子亚型的具体步骤如下。
1.2.1 筛选正常组织样本中基因间相对表达秩次关系稳定的基因对将背景基因两两组合,如果一对基因(基因i 和基因j)在正常样本内满足P(Gi>Gj)≥0.9[公式⑴],那么该基因对定义为稳定基因对。
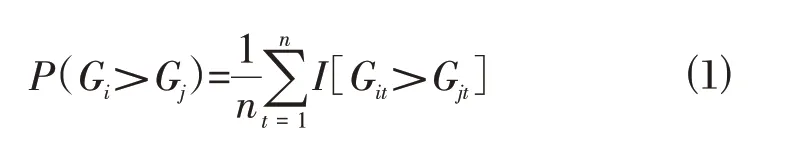
其中n表示所有的正常样本数,t代表第t个样本,Gi>Gj表示基因i和基因j的表达秩次关系。
1.2.2 基于稳定基因对筛选疾病组织样本中的显著逆转基因对基于正常样本中得到的稳定基因对,评估其在疾病中相对表达秩次关系改变情况。对于任一稳定对(Gi,Gj),假设在正常样本中观察到Gi>Gj的样本有n1个、Gi≤Gj的样本有n2个,在疾病样本中满足Gi>Gj的样本有m1个、Gi≤Gj的样本有m2个。利用Fisher 精确检验,可检验Gi、Gj间相对表达秩次关系在正常和疾病样本间分布是否存在差异。经多重假设检验校正后,满足假阳性发现率(FDR)<5%的基因对定义为逆转基因对。
1.2.3 筛选具有潜在疾病分子亚型鉴别能力的基因对对于在疾病中筛选得到的逆转基因对,按如下步骤进一步筛选具有潜在疾病分子亚型鉴别能力的基因对:⑴逆转对的秩次关系的改变只在20%~80%的疾病患者之中发生。这是为了确保基因对具有鉴别疾病分子亚型的能力。⑵对于任意逆转对(Gi,Gj),根据其相对表达秩次关系,可将疾病样本分为两组,Gi>Gj为1 组,Gi≤Gj为1 组。然后利用t检验,控制FDR 为5%,识别两组间的差异基因,并基于KEGG(京都基因与基因组百科全书,https://www. kegg. jp/)通路数据库对两组间识别的差异基因进行功能富集分析。若一个生物学通路显著富集了差异基因,则认为该通路发生了生物学功能的扰动。对于一个逆转基因对,可按其在疾病样本中的相对表达秩次关系将疾病样本分成两组。如果这两组样本间存在差异表达基因,同时存在生物学功能的扰动,则认为该基因对具有鉴别潜在疾病分子亚型的能力,称其为分子亚型候选基因对。
1.2.4 分子亚型识别首先,按照分子亚型候选基因对扰动通路的数目,将基因对进行降序排列。然后,构建一个分子亚型候选基因对×疾病样本的矩阵,其中元素rtij为矩阵中第t个样本中基因对(i,j)的相对秩次关系,其值为1 或0(如果Gi>Gj,则为1,如果Gi≤Gj,则为0)。以1 000 为梯度,分别取前k个基因对(k=1 000*n,n=1,2,3…)对应的矩阵,利用欧式距离进行聚类分析,识别疾病分子亚型。为了保证结果的可靠性,对k个基因对聚类结果,利用随机实验进行验证:随机从所有分子亚型候选基因对中挑选k 个基因对进行聚类分析,重复100 次。如果随机分类结果与前k个基因对的分类结果一致率<95%,则重新选择标记。反之,将其作为分子亚型预测标记,用作后续分析。
1.3 基于加权基因共表达网络识别疾病分子亚型相关基因模块基于加权基因共表达网络(WGCNA)分析提取与疾病亚型显著相关的模块(用R 包WGCNA 实现)[13]。首先,对背景基因表达矩阵相关系数进行加权,使基因间的相互作用关系符合无标度分布。然后对基因进行分类,并将具有相似表达模式的基因分成一个模块,最小模块大小(min-ModuleSize)设置为100,其他参数设置为默认值。同一模块的基因往往表现出相似的表达模式和功能[14]。
1.4 模块基因功能富集分析基于京都基因和基因组百科全书(KEGG)数据库对模块基因进行通路富集分析,用R包clusterProfile实现[15]。
1.5 PPI 网络构建和核心基因筛选基于蛋白质互作在线数据库STRING(www. string-db. org)对候选模块内的基因进行蛋白与蛋白之间的互作分析(PPI),利用网络最大集团度(MCC)评分算法[公式⑵][16],识别PPI网络中的关键基因。
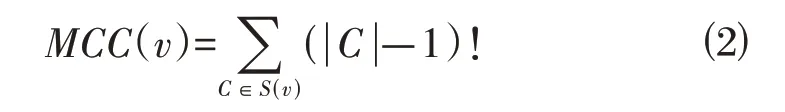
其中,集合S表示集合中元素的数量,S(ν)是包含ν 的最大集团的集合,(|C|-1)!是所有小于|C|的正整数的乘积。
2 结 果
2.1 具有潜在疾病分子亚型鉴别能力的基因对按照方法学筛选具有潜在疾病分子亚型鉴别能力的基因对流程。基于数据集GSE46960,在每个样本中分别对基因的表达水平进行两两比较,形成基因对×样本的0-1 矩阵X。Xij表示第i 个基因对(Ga,Gb)在第j 个样本中的相对表达秩次关系,其值为1或0(1代表Ga>Gb,0代表Ga≤Gb)。在7例正常婴儿样本中,有144 822 185个基因对在90%以上的正常样本组织中具有Ga>Gb的表达模式,其中有5 105 085个基因对的相对表达秩次关系在BA 样本中发生显著逆转Ga≤Gb(Fisher 精确检验,FDR<0.05),暗示了这些基因对的相对表达秩次关系可能与BA 的发生发展相关。对每一个逆转基因对,进一步根据其在疾病样本中的相对表达秩次关系,将疾病样本分为两组(Ga>Gb为一组,Ga≤Gb为一组),然后识别两组间的差异基因并基于KEGG数据库进行功能富集分析,发现有4 836 202 个基因对满足条件,用作后续分析。
2.2 识别BA 的分子亚型基于亚型候选基因对,首先构建亚型候选基因对×疾病样本的0-1 矩阵X。矩阵中xij表示第i 个基因对在第j 个样本中的相对表达秩次关系,其值为1 或0(1 代表>,0 代表≤)。按照亚型候选基因对富集到的生物学功能通路个数由大到小进行排序,然后分别选取前1 000 对、2 000 对、3 000 对、4 000 对和5 000 对基因对,对64例BA 样本分别进行聚类分析。结果显示前3 000 2A),采用动态剪切法划分模块,合并相似度>75%的模块,最终构建了17 个共表达模块(图2B)。进一步分析了BA 不同亚型与各种基因模块的相关性,发现洋红色(Magenta)模块与BA 亚型最相关(|r|=0.58,P=6×10-7)(图2C)。
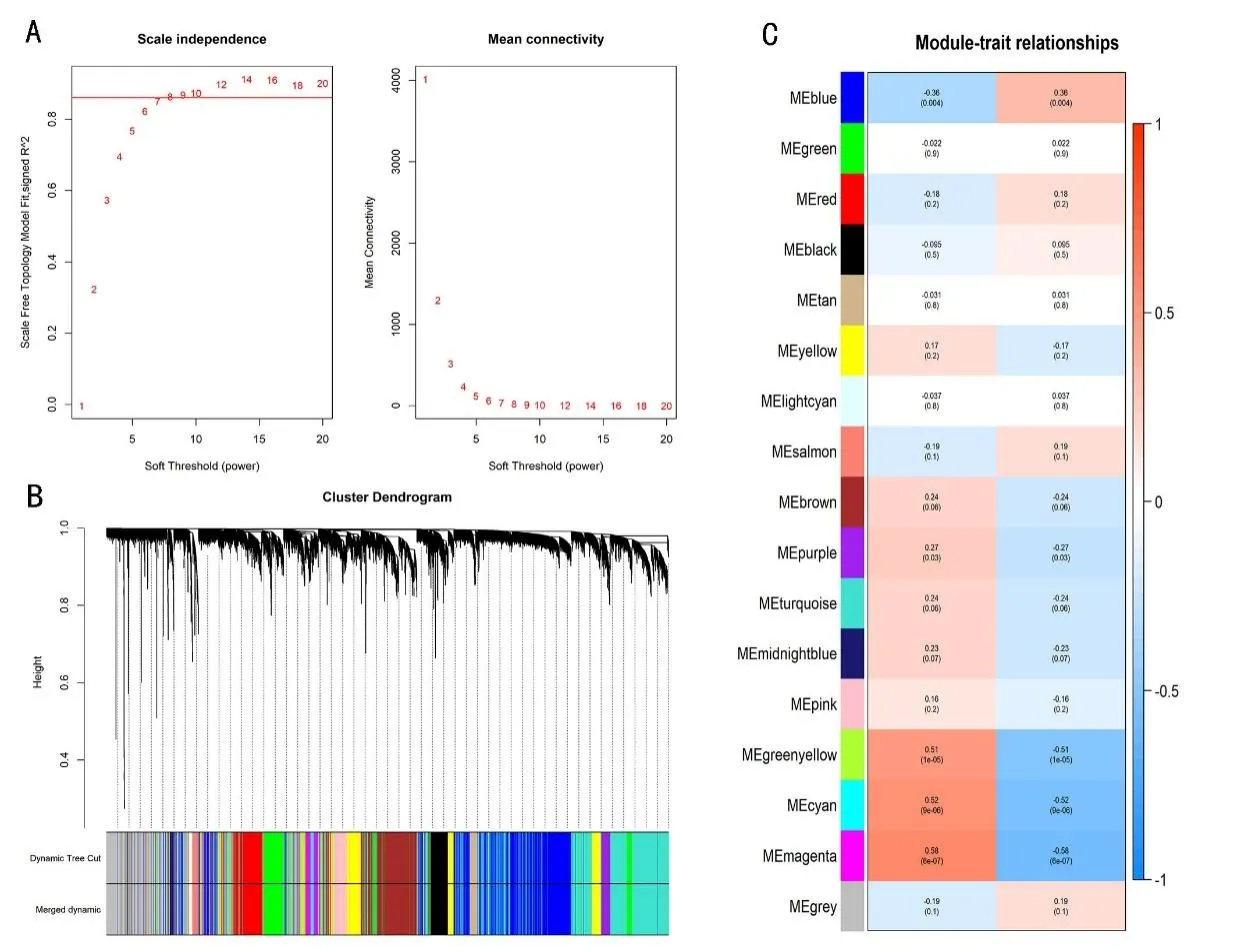
图2 BA亚型基因共表达网络模块的构建
基于KEGG数据库,对Magenta模块中涉及的基因进行富集分析,发现这些基因主要涉及PI3K-Akt信号通路、黏附斑激酶通路(FAK)和ECM 受体相互作用通路等(图3A)。对、4 000 对、5 000 对的聚类结果与随机情况一致。因此,本文将前3 000 对基因对的聚类结果作为BA分子亚型识别依据。聚类结果显示64例BA 样本明显聚集为两类:一类样本个数为54 个,另一类样本个数为10个(图1A)。
为了进一步验证BA 中存在的不同分子亚型,我们把得到的前3 000 个亚型候选基因对应用于GSE15235 数 据集中,GSE15235 数据集中43 例BA样本中依然明显聚集为两大类:一类样本个数15个,另一类样本个数为28 个(图1B)。通过数据集GSE46960、GSE15235 间两类样本间的差异基因比较发现,在GSE46960 两类间识别了436 个差异基因,GSE15235 两类间识别了584 个差异基因,共交叠了75个差异基因(超几何分布P=4.8×10-32),并且这75 个差异基因有69 个基因在两组间失调方向一致(二项分布P=5.77×10-15)。
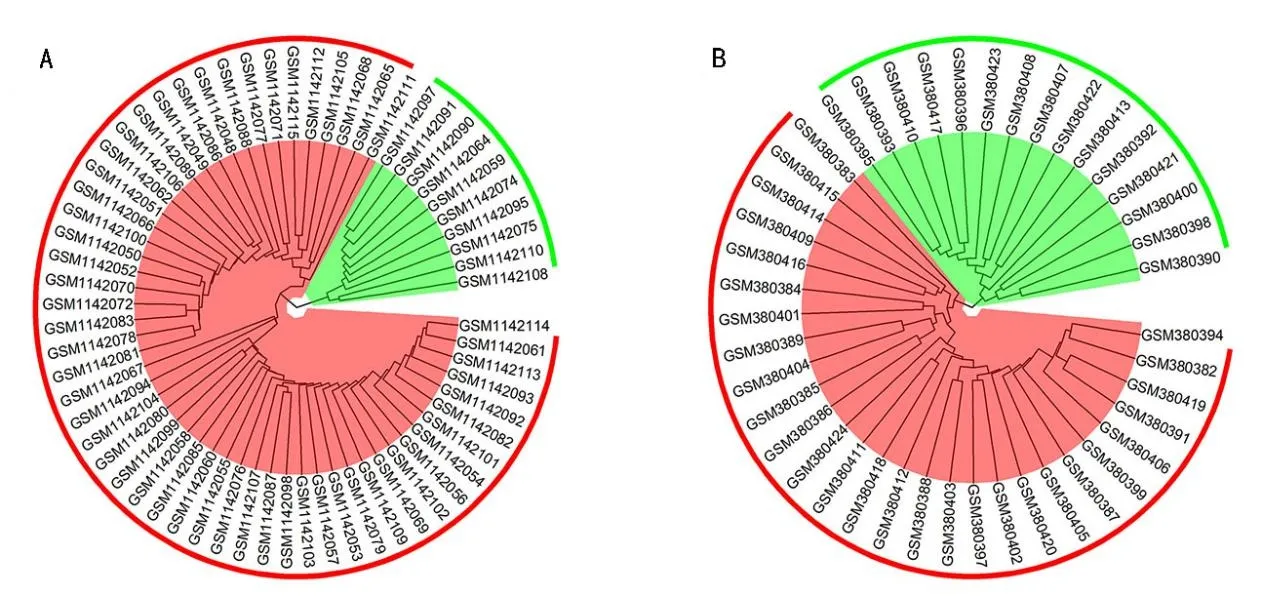
图1 BA样本分子亚型分类
2.3 BA 疾病亚型共表达模块构建及核心基因识别为了进一步分析BA 中不同亚型的基因表达模式,利用WGCNA 方法对数据集GSE46960构建加权共表达网络。使用无标度拓扑标准选择β=8(图
将所有Magenta 模块内的基因导入STRING 数据库以构建PPI 网络,通过MCC 算法发现了10 个BA 亚型相关的核心基因(LUM、COL6A3、FBN1、SPARC、DCN、LAMA4、FAP、ANTXR1、LAMA2 和COL1A2)。LAMA2、LAMA4 和LUM 基因(图中红色基因)与其他蛋白质的相互作用更频繁(图3B)。

图3 Magenta模块中基因的KEGG通路富集分析和PPI网络构建
对10个核心基因在GSE46960数据集的正常组织、BA亚型Ⅰ和亚型Ⅱ三组间的表达情况进行分析发现,相对于正常组织,10 个基因在亚型Ⅰ中显著高表达,而在亚型Ⅱ中显著低表达(图4A)。在GSE15235 数据集中,我们还发现这10 个核心基因的表达水平在纤维化和炎症两类样本间有显著差异,且纤维化组的表达水平显著高于炎症组(图4B),与BA亚型Ⅰ和亚型Ⅱ一致。
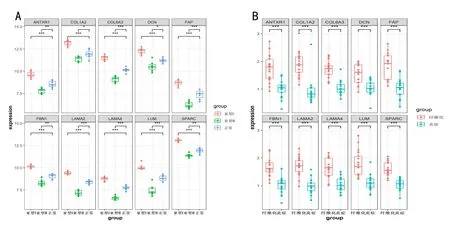
图4 10个核心基因在不同亚型间的表达分布情况
3 讨 论
本研究基于样本内基因间相对表达秩次关系,开发了疾病分子亚型识别算法,确定了在BA 疾病中存在两组不同的疾病亚型,并利用WGCNA 分析筛选出了与亚型相关共表达基因模块。通路富集分析显示,Magenta 模块内基因主要涉及PI3K-Akt信号通路、黏附斑激酶通路(FAK)和ECM 受体相互作用通路等。有研究表明,PI3K-Akt 信号通路可能抑制NF-κB 和NLRP3 炎症通路的蛋白表达,从而减少炎症因子的分泌[17]。FAK 和ECM 受体相互作用通路抑制TGF-β1信号传导,降低成纤维细胞分化[18]。最后,本文还识别了与BA 分子亚型相关的10 个核心基因。其中,LAMA2 和LAMA4 可促进转化生长因子(TGF-β)信号传导,而TGF-β 信号会阻止慢性损伤期间促纤维化FAP 的细胞凋亡,从而促进纤维化环境[19-20]。KRISHNAN A 等[21]研究表明,LUM 可在体外诱导生长因子β1(TGF-β1)转化为促纤维化细胞因子。COL1A2和COL6A3编码胶原蛋白,而胶原蛋白的过度表达是肝纤维化的基本特征。当肝星形细胞(HSCs)被激活时,基质金属蛋白(MMPs)的表达增加,同时MMPs 的抑制剂基质金属蛋白酶(TIMPs)的表达增加。如果TIMPs 增加过快,MMPs/TIMPs 的比值会发生变化,使细胞外基质(ECM)的合成和降解失衡,促进纤维化的发展[22-23]。其余5个关键基因(FBN1、SPARC、DCN、FAP 和ANTXR1)虽然还未有其与BA 相关报道,但其参与了蛋白质消化吸收、ECM-受体相互作用和PI3K-Akt 信号通路。其中ECM 的变化由多种介质和生长因子诱导,从而调节各种效应,如刺激血管生成、炎症反应和促进基质侵袭,可导致成纤维细胞的迁移或分化,最终使细胞纤维化[24-25]。提示这10 个核心基因与BA 进行性肝炎、肝纤维化有关。
在本研究中,正常样本相对较少,在稳定基因对筛选过程中,可能引入一定的假阳性基因对,后续需要更多的正常样本来验证。同时,本研究筛选的亚型候选基因对数量较多,不利于后续聚类鉴别疾病亚型。因此,如何筛选出更少、更具有生物学意义的亚型候选基因对是未来的研究重点。后续研究我们将考虑如基因在候选对中出现的频率、基因间的表达相关性等因素进一步改进算法。
