利用连接酶检测反应检测碎片化新冠病毒S基因D614G突变
2022-06-18张新亚罗志丹
张新亚,张 建,卢 辰,薛 勇,罗志丹
(1.江苏海洋大学 江苏省海洋药物活性分子筛选重点实验室,连云港 222005;2.江苏省海洋资源开发研究院(连云港),连云港 222005)
新型冠状病毒(SARS-CoV-2)所引发的新型冠状病毒肺炎(COVID-19)是世界卫生组织认定的全球大流行传染病[1-3],具有传播速度快、感染范围广、防控难度大等特点,迄今仍在全球不断蔓延,防控形势依然十分严峻[4-6]。新冠病毒属于单链正义RNA病毒,在复制过程中容易发生突变,目前研究者已经收集了200多万个新冠病毒基因组序列。尽管绝大多数基因突变都不会影响病毒的毒性和传染性,但部分新冠病毒基因组上第23 403位的腺苷酸突变为鸟苷酸,导致新冠病毒S蛋白第614位的天冬氨酸变为甘氨酸,这一突变可以增强蛋白酶对S蛋白的切割能力,从而促使病毒具有更强的感染能力[7]。此外,D614G突变改变了S蛋白甚至是整个病毒的免疫原性,进而降低了康复期血清对病毒的敏感性。携带该突变的新冠病毒毒株自2020年2月在欧洲出现后,很快成为全球主要流行毒株之一,并进一步演化出德尔塔等突变株。因此,如何在病毒核酸检测时快速鉴定这些单核苷酸多态性(SNP),一直是研究者关注的重要问题。
目前主流的核酸单个SNP检测手段包括测序法和荧光定量PCR法[8-10]。其中测序法最为准确,但耗时较长,不利于临床快速检测;荧光定量PCR法检测RNA突变[11-14],首先需要将模板RNA逆转录为cDNA,然后使用一对根据突变位点上下游设计的PCR引物,以及一条覆盖突变位点的Taqman-MGB荧光探针进行荧光定量PCR,该方法需要的模板长度一般不低于100 nt,如果病毒核酸在提取和保存过程中受到破坏,碎片化程度较严重,则会严重影响检出率。
连接酶检测反应(ligase detection reaction, LDR)是一种不依赖聚合反应的检测技术[15-17]。其原理为利用DNA连接酶催化两条寡核苷酸单链之间形成磷酸二酯键时,要求缺口处的核苷酸必须形成互补双链的特性,设计特异性连接探针。该方法直接针对SNP位点上下游设计探针,当探针与SNP位点完全匹配的时候才能在连接酶作用下进行连接,连接产物可通过各种方法进行检测。理论上LDR可检测的片段长度只取决于探针与模板的配对区域长度,可以低到50 nt甚至更短。研究期望利用LDR技术,开发一种快速检测碎片化新冠病毒S基因D614G突变的方法,为病原核酸检测开辟新的思路。
1 材料和方法
1.1 材料
新冠病毒S基因质粒购自通用生物系统(安徽)有限公司;TaqSYBR Green qPCR Premix、无缝克隆试剂盒和DNase I购自江苏愚公生命科技有限公司;HiFiTaqDNA连接酶、T7 RNA聚合酶购自NEB。
1.2 方法
1.2.1 设计引物探针
利用连接酶反应特性,针对突变位点两侧序列设计一对特异性的单链DNA探针。其中下游探针的5′部分与目标突变位点上游序列反向互补,5′末端带有自由磷酸基团,3′部分为通用序列;上游探针的5′部分为通用序列,3′部分与目标突变后位点及其下游序列反向互补,其中3′末端碱基与突变后位点碱基互补(图1)。然后设计一组PCR引物与探针的通用序列配对。
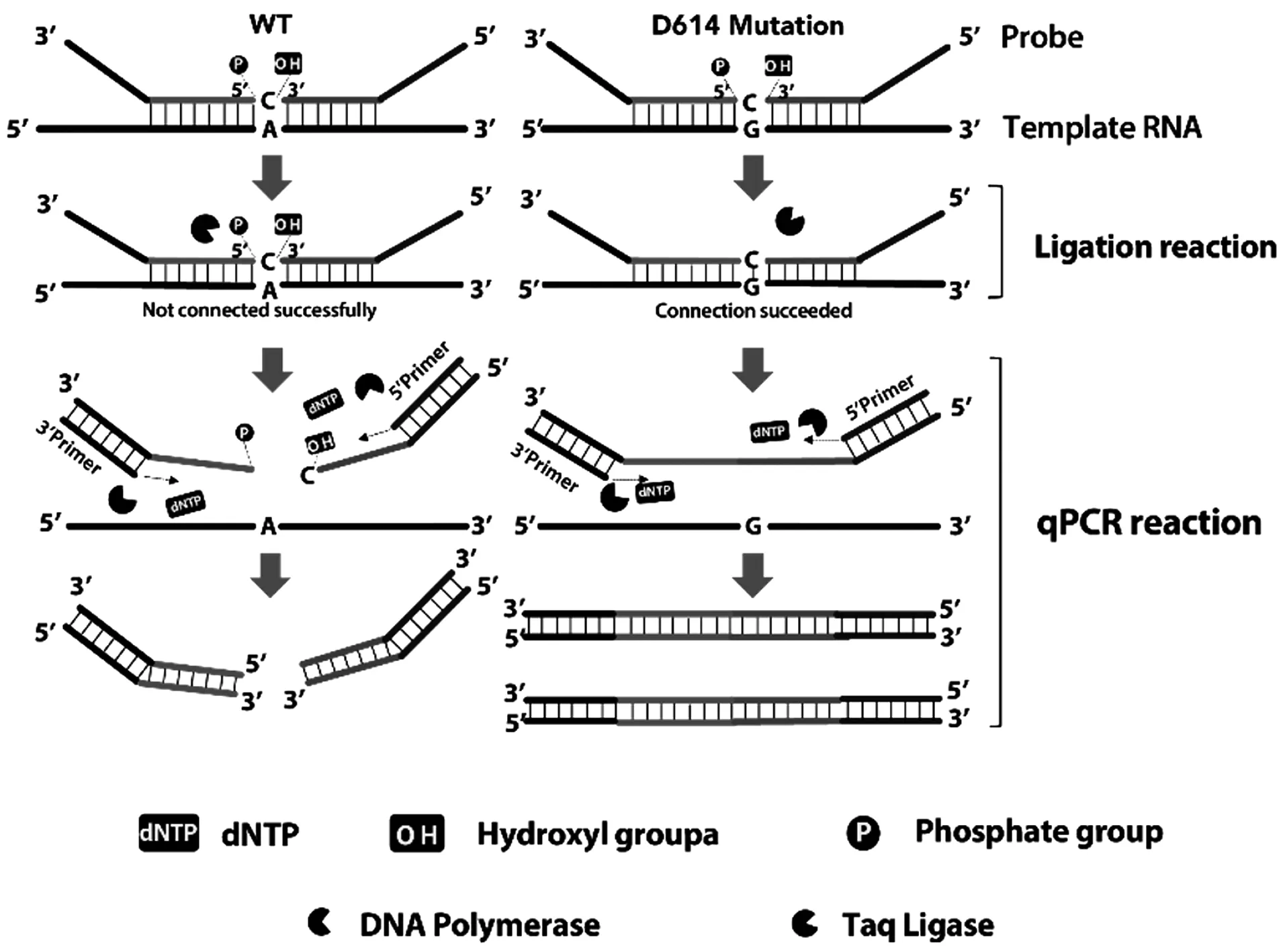
图1 LDR-qPCR原理图
1.2.2 获取新冠病毒S基因RNA片段模板
以带有野生型新冠病毒S基因的质粒为模板,使用无缝克隆技术人工构建D614G突变体。根据D614G突变位点的上下游合计50、100和200 bp的序列设计一组产物长度不同,但均带有T7启动子的引物,对野生型和突变体质粒进行PCR扩增。以纯化后的PCR产物为模板,使用T7 RNA聚合酶进行体外转录获得长度不同的野生型和D614G突变体病毒RNA片段,经DNase I消化去除DNA后,使用琼脂糖凝胶电泳验证。
1.2.3 建立LDR-qPCR反应体系
使用体外转录获得的新冠病毒S基因RNA片段为模板,配制表1所示的反应体系进行LDR反应,按照HiFiTaqDNA连接酶说明书推荐的程序:95 ℃预变性3 min;95 ℃变性30 s,58 ℃退火连接4 min,循环35次。随后使用PCR引物对LDR产物进行荧光定量PCR检测。

表1 LDR反应体系
首先筛选LDR模板浓度,分别设置转录后原液、稀释10、100倍等3个梯度;随后筛选探针浓度,设置为0.1 μmol/L和0.01 μmol/L两个梯度;然后筛选最适的探针与模板配对区长度,分别设置25、22、18、15、12和8 bp。筛选出较优条件后,测试LDR-qPCR检测不同长度病毒RNA片段的表现,片段长度设置为200、100和50 nt。
1.2.4 模拟样本测试
提取Hela细胞总RNA;同时以带有新冠病毒N基因的质粒为模板进行体外转录获得N基因RNA,并稀释到与S基因RNA片段相同浓度。将不同浓度的S基因D614G与野生型模板分别与细胞总RNA、N基因RNA混合,模拟真实提取的组织RNA。使用LDR-qPCR体系进行检测。
1.2.5 测试探针与模板不完全匹配检测效果
以S基因D614G突变位点为中心,设计长度为50 nt,向上下游各移动5~10 nt、与探针不完全匹配的一组RNA片段(图2),使用上述的LDR-qPCR体系验证检测效果。
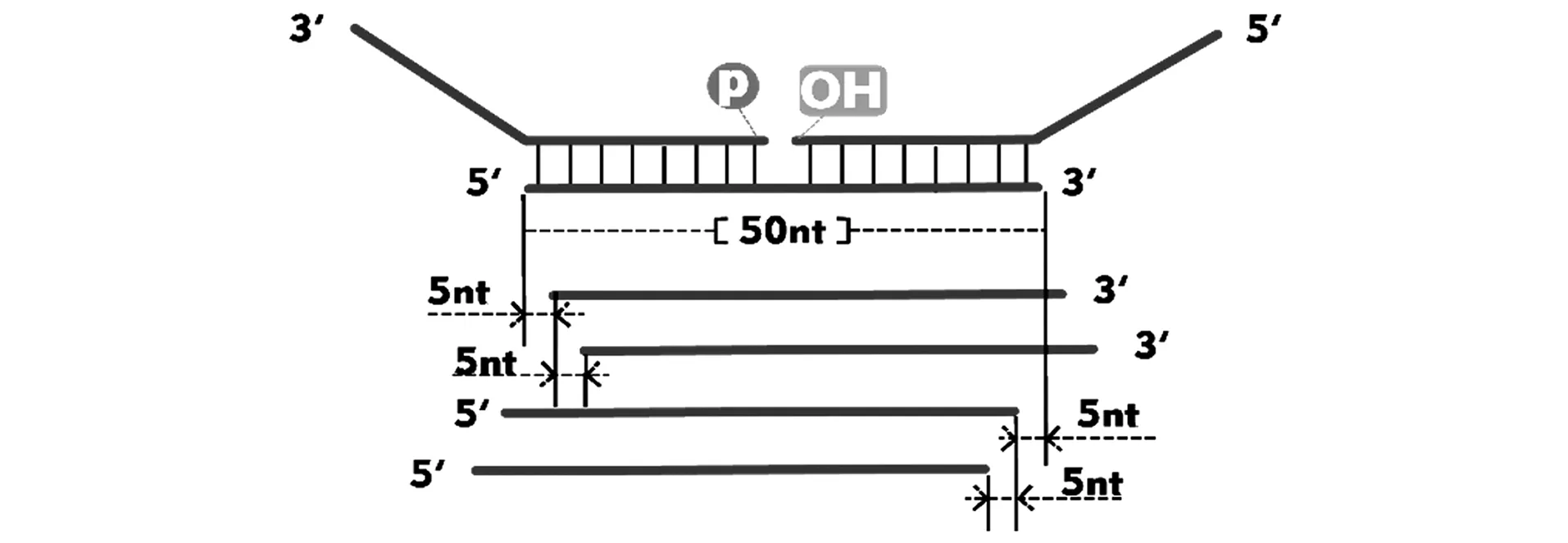
图2 探针错配原理图
2 结果与分析
2.1 新冠病毒S基因RNA的获取
以带有野生型新冠病毒S基因的质粒为模板,采用无缝克隆技术构建D614G突变体,经测序验证正确。分别以野生型和突变体质粒为模板,使用不同引物对扩增出含有S基因614位点,长度为200 bp、100 bp和50 bp的片段。经T7 RNA聚合酶体外转录,成功获得长度为200 nt、100 nt和50 nt的新冠病毒S基因RNA片段(图3)。
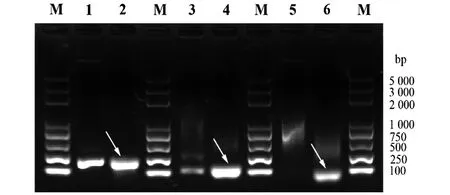
M:DL2000 Plus Marker;泳道1、2为长度200 nt,转录前后片段;泳道3、4为长度100 nt,转录前后片段;泳道5、6为长度50 nt,转录前后片段。箭头指示所获得的RNA片段。
2.2 RNA模板与探针浓度筛选
长度为200 nt的野生型和D614G突变体S基因片段体外转录后RNA浓度均为50 pmol/L,取原液、10倍稀释和100倍稀释的RNA分别进行LDR-qPCR检测,结果表明对50 pmol/L和5 pmol/L浓度的模板,突变体的Ct值显著低于野生型,具有良好区分度;而对0.5 pmol/L的模板,尽管突变体和野生型Ct值之间也具有显著性差异,但与5 pmol/L相比差值偏小,且突变体Ct值偏高[图4(a)]。因此,以5 pmol/L的RNA作为后续实验的模板浓度。比较工作浓度为0.1 μmol/L和0.01 μmol/L的探针检测表现,结果表明两个浓度的探针均有较好的区分效果[图4(b)]。
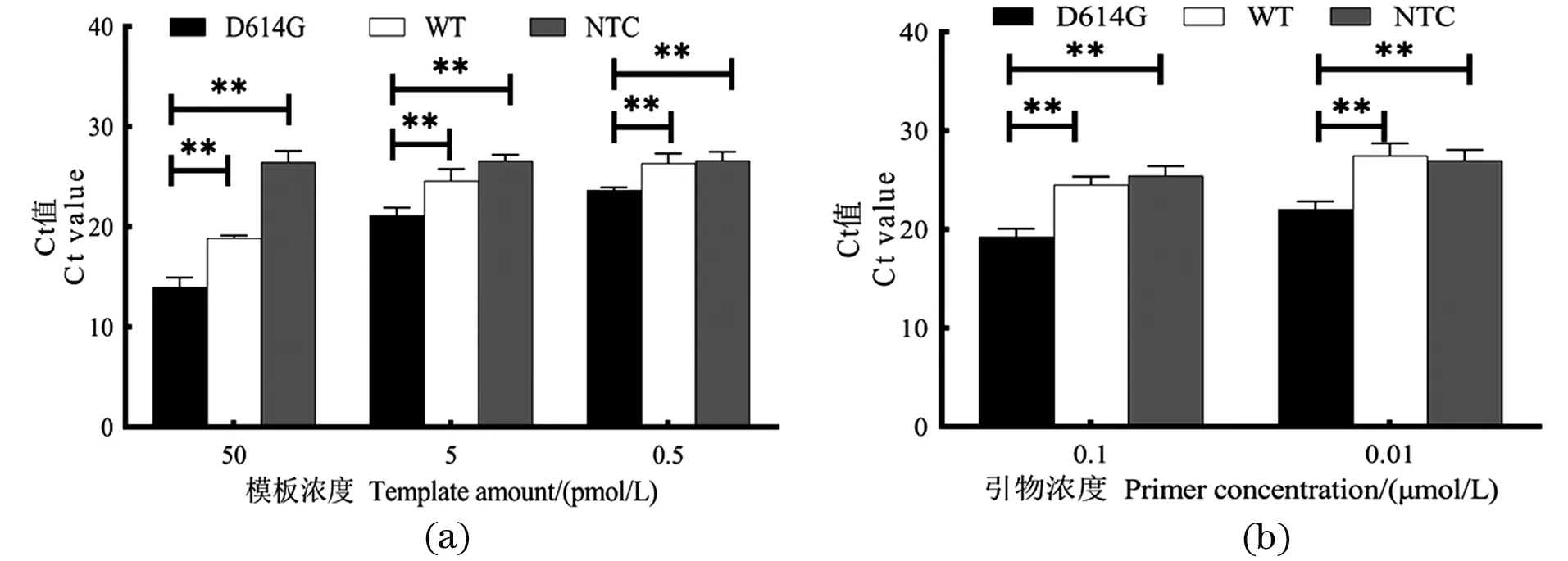
(a)RNA模板浓度筛选;(b)LDR探针浓度筛选。** 表示差异达到显著水平(P<0.01)。
2.3 探针模板配对区长度筛选
LDR探针由模板配对区和通用扩增区两部分组成,容易形成引物二聚体从而干扰qPCR结果,因此需要测试探针中模板配对区长度对结果的影响。每一侧探针都分别设置15、18、22和25 nt的模板配对区,分别对野生型和突变体模板进行检测。结果表明,包含有18、22、25 nt模板配对区的探针,对D614G突变体有良好的区分度(图5)。后续实验中继续使用具有25 nt模板配对区的LDR探针。
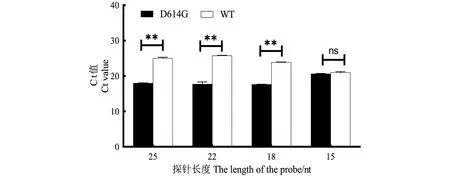
** 表示差异达到显著水平(P<0.01);ns表示无显著性。
2.4 不同长度RNA模板检测
LDR体系为检测碎片化片段提供了独特的优势,理论上其可检测片段长度只取决于上下游探针的模板配对区长度之和。围绕D614G位点上下游构建了200、100和50 nt等3种长度的病毒RNA片段模板,用同样的探针进行了测试。结果表明,3种长度的突变体Ct值均显著低于突变体(图6),表明本方法至少可以检测到50 nt片段中所包含的点突变,而普通荧光定量PCR对这个长度的模板是无能为力的。
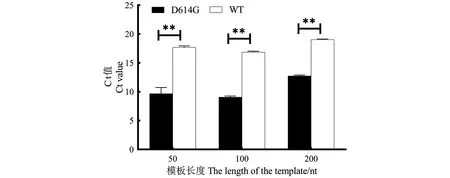
** 表示差异达到显著水平(P<0.01)。
2.5 模拟真实提取样本检测
实验室不具备操作高危病原微生物的条件,因此将提取的细胞总RNA(3 000 ng/μL)、体外转录获得的新冠病毒N基因RNA与S基因片段混合,模拟提取的患者样本来验证本方法的分辨率。结果表明,当5 pmol/L的S基因RNA混入到其他RNA中时,突变体和野生型的Ct值仍然保留显著差异(图7),说明方法具有足够的分辨率。
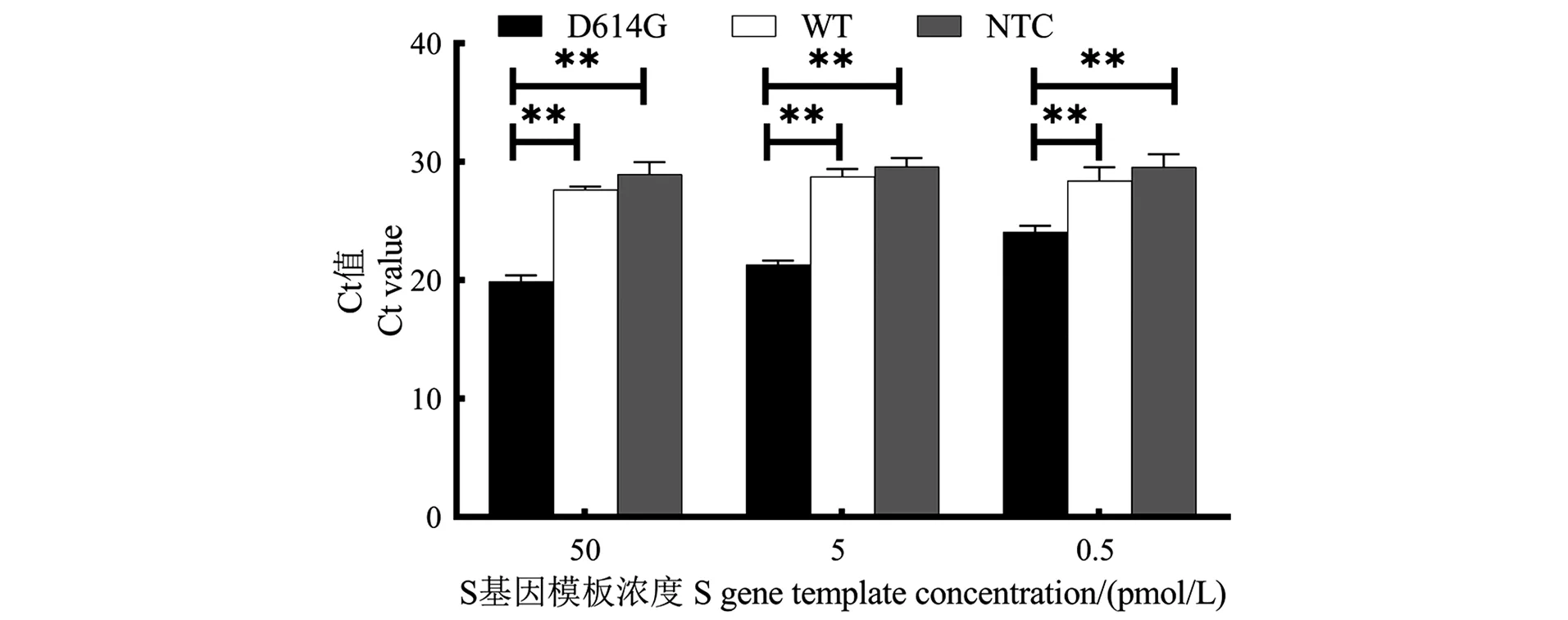
** 表示差异达到显著水平(P<0.01)。
2.6 探针-模板不完全匹配测试
上述实验所模拟的是较为理想的情况,即LDR探针的模板配对区恰好可以与病毒RNA片段完全匹配。但事实上,碎片化的病毒RNA片段往往还不足以与探针恰好完全匹配。为了模拟这种情况,设计了一组与探针不完全匹配的病毒RNA片段(图8)来验证本方法的可行性。结果表明,即使模板与LDR探针错位达到10 nt,突变体和野生型的Ct值依然有6~8个循环的差别,足以进行区分。换言之,检测长度下限至少为40 nt。
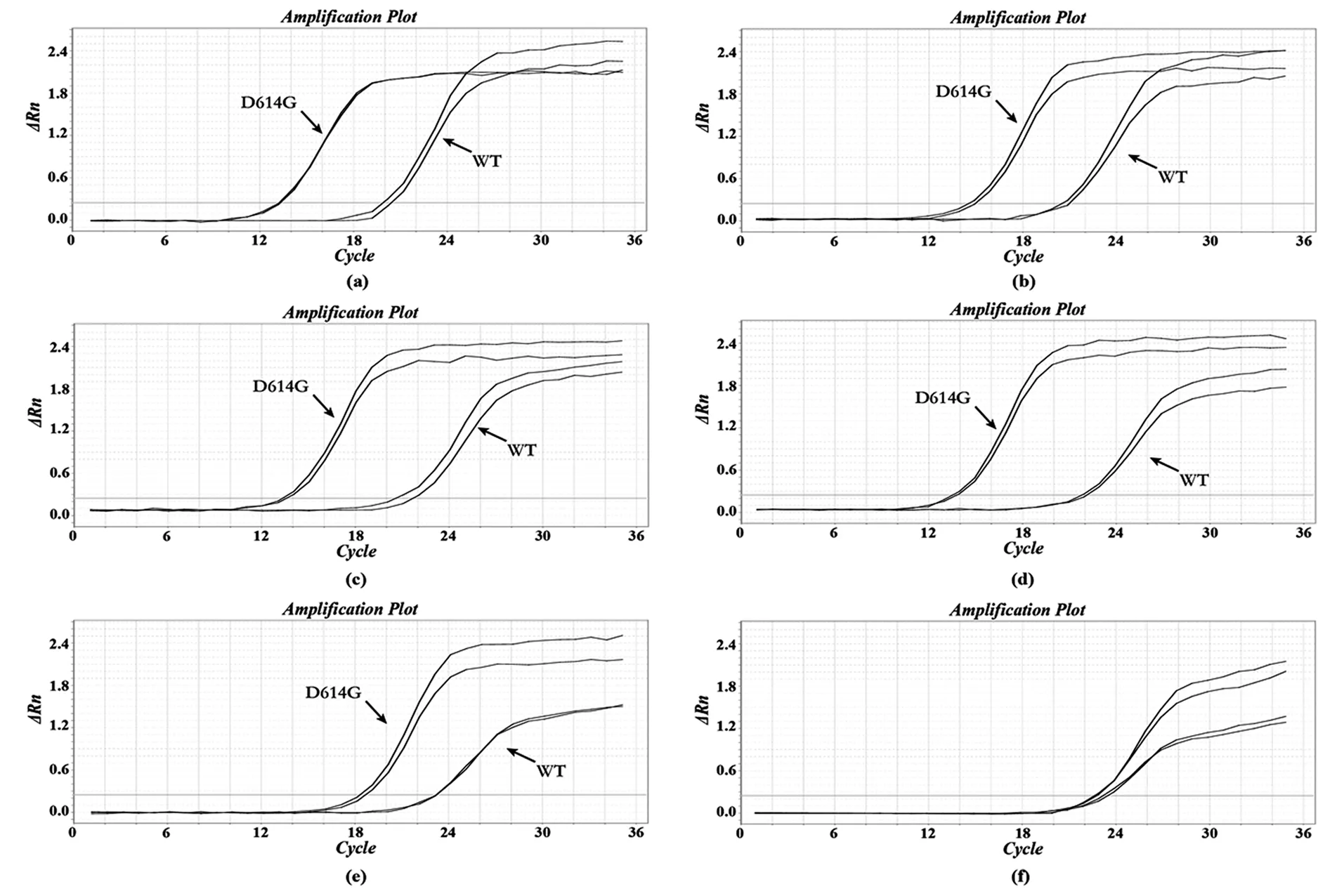
(a)模板区50 nt与探针区(50 nt)完全匹配;(b)模板区50 nt相对探针区(50 nt)向上游移动5 nt;(c)模板区50 nt相对探针区(50 nt)向上游移动10 nt;(d)模板区50 nt与相对探针区(50 nt)右移向下游移动5 nt;(e)模板区50 nt与相对探针区(50 nt)向下游移动10 nt;(f)NTC(不加模板)。
3 讨论
目前主流的SNP快速检测方法使用的Taqman-MGB探针荧光定量PCR法,需要考虑SNP位点在探针上的位置和附近的序列特征,如果SNP位点附近存在特殊结构,则可能无法设计出有效的探针;探针必须与引物相隔一定距离,所需模板长度一般要求不低于100 nt,在检测碎片化病毒基因时受到限制。而LDR技术直接针对SNP位点侧翼序列设计探针,无需考虑序列特征,设计简单方便;而且可检测片段长度理论上只取决于上下游探针的模板配对区长度之和,可以检测的片段长度可以低于50 nt。研究建立的方法所需模板最低长度仅为40 nt,远低于普通探针法荧光定量PCR所需的模板长度,在检测碎片化模板上具有较大优势。
目前LDR反应第一步依赖探针和模板之间的自由结合,效率相对偏低,而且连接酶说明书所用循环反应程序是针对双链DNA模板设置的,未针对单链RNA模板进行优化,后续还需要进一步优化。此外,研究检测LDR产物使用荧光定量PCR,操作较为繁琐,而且LDR探针上额外加入荧光定量PCR扩增所需通用序列,使得探针长度偏长,容易产生引物二聚体影响检测效果。后续可以考虑采用其他检测手段加以改进。
