基于理论计算探究CYP4F12催化花生四烯酸机制
2022-06-18吕旭东陶玉莲马宇飞张美玲
吕旭东,陶玉莲,马宇飞,颜 菲,张美玲
(天津医科大学 生物医学工程与技术学院,天津 300070)
细胞色素P450(CYP)同工酶是一类含血红素的单加氧酶家族,主要位于线粒体内膜或真核细胞内质网膜中[1]。人类基因组中编码有57种CYP蛋白,这些基因被分成18 个家族和43 个亚家族。CYP的精确命名由CYP(细胞色素的正式缩写)、家族(数字)、亚家族(字母)和同工型(数字)按顺序书写。通常,CYP家族1、2和3包含主要的异种生物代谢酶,在药物基因组学风险中起主要作用,而CYP4酶参与脂肪酸的代谢,与遗传疾病风险相关[2]。CYP4F12是 CYP4酶中目前研究较少的药物代谢酶,主要在肝脏、肾脏、肠道中表达[3-4]。对肝癌中CYP4表达谱的评估表明CYP4F12是肝癌预后的有利因素[5]。据相关研究表明,CYP4F12可以催化花生四烯酸(AA)发生羟基化反应[6]。AA及其代谢物对肿瘤的进展以及血管和肾脏的血管生成和血压调节具有重要作用[7]。目前,并没有相关报道解释CYP4F12催化AA的机制。
细胞色素P450依赖性ω-羟基化是CYP4家族成员的原型代谢反应,对治疗药物、内源性化合物的消除和生物活化都很重要[8]。这种ω-区域选择性不是绝对的,并且大多数CYP4酶会产生末端链羟基化产物的混合物。羟基化产物ω:ω-1,ω-2,ω-3的比率范围可以从>20∶1到0∶1,变化范围很大[9]。CYP4F8、CYP4F12、CYP4X1和CYP4Z1都不是严格ω羟化酶,这些酶的底物特异性和产物区域选择性尚未恰当定义。
为了解底物反应性和活性位点残基在CYP4酶介导的ω-羟基化中的作用,本文研究CYP4F12介导AA内部羟基化作用。将密度泛函理论(DFT)计算与分子对接,分子动力学(MD)模拟和结合自由能(MM/GBSA)计算一起应用于解释CYP4F12催化AA的代谢。这些方法的组合有助于了解复杂系统(如AA)的酶代谢过程,并正确解释CYP4F12催化AA的机制。
1 材料与方法
1.1 DFT计算
研究建立了CYP的Compound I(Cpd I)模型,Cpd I是血红素的最终氧化态形式,如图1(a)所示。计算在B3LYP水平上进行。几何优化中,Fe原子使用LANL2DZ赝势基组,其他原子使用6-311+G(d,p)基组。使用Becke-Johnson阻尼考虑色散作用。最终能量计算中使用隐式连续介质模型(COSMO)考虑溶剂效应。使用零点振动能(ZPE)进行能量校正,温度设置为298.15 K。使用频率计算验证得到的过渡态。所有计算使用Gaussian 16软件包[10]进行。

(a)反应路径图;(b)底物潜在代谢位点。
1.2 蛋白3D模型的构建
在PSI-BLAST(https://www.ebi.ac.uk/Tools/sss/psiblast/)上基于CYP4F12序列搜索模板。在综合考虑下选择得分最高的6C93、5T6Q、6MA7、3TJS蛋白用于多模板建模。使用Modeller 9软件包[11]生成CYP4F12模型。血红素分子从模板蛋白6C93[12]获取并定位于建模蛋白。最终使用Verify3D[13]、ERRAT[14]、PROCHECK[15]和QMEAN[16]程序评估模型质量。
1.3 分子对接
分子对接被广泛用于生物分子相互作用和机理的研究[17]。本文使用AutoDock 4.2软件[18]进行对接。AA结构来自Pubchem库(https://pubchem.ncbi.nlm.nih.gov/)经DFT方法优化得到。对接使用60×60×60网格的三维周期性对接盒子,以AA为对接中心,格点间距为0.0375 nm。蛋白设置为刚性,配体为柔性。生成的构象数为20。
1.4 分子动力学模拟
使用Gromacs 5.1.4软件[19]来MD模拟。模拟中使用charmm 36 力场[20],小分子的charmm力场参数在CHARMM-GUI服务器(http://www.charmm-gui.org)上生成。模拟在TIP3P水溶剂模型及十二面体盒子下进行,蛋白到盒子边缘的距离为1.4 nm。添加3个Na+以保持系统中性。能量最小化的方法为最陡下降法,当能量小于1 000.0 kJ/mol时进行收敛。在1 ns的NVT系综和5 ns的NPT系综的预平衡后模拟体系稳定。最后体系进行500 ns的MD模拟。时间步长为2 fs,每2 ps保存一次轨迹。
2 结果与分析
2.1 DFT预测羟基化的区域选择性
运用DFT理论研究Cpd I模型系统[图1(a)]是探究CYP酶在药物代谢中机制的有用方法[21]。AA有几种可能的代谢位点会发生羟基化:ω(ω′,ω″,ω‴),ω-1(ω-1R,ω-1S),ω-2(ω-2R和ω-2S),ω-3(ω-3R和ω-3S)和ω-4(ω-4R和ω-4S)见图1(b)。脂族羟基化速率决定步骤是Cpd I介导的氢提取反应。
表1列出各个代谢位点提取氢的活化势垒,它们均在DFT计算的能量的正常范围内。计算预测活化势垒最高的是ω位点,范围为77.25~81.06 kJ/mol;而活化势垒最低的位于ω-2位点,值为55.34 kJ/mol。先前的实验结果表明[22],CYP4F2催化的AA主要生成ω-2羟基化产物。因此,DFT计算结果与实验观察结果一致。

表1 潜在催化位点的反应能垒
2.2 人类CYP4F12同源模型的产生和验证
CYP4F12建模蛋白(图2)与模板序列的全同率(sequence identity)为44.89%。在TMHMM v.2.0(http://www.cbs.dtu.dk/services/TMHMM/)服务器上对CYP4F12序列进行跨膜预测,删除构成膜嵌入结构域的前37个残基,这些残基远离活性位点且不会影响模型构建。

(a)建模蛋白正面图;(b)建模蛋白背面图;(c)建模单板(青)与模板(灰)叠加。
选取moddller程序输出DOPE得分最佳的模型用于后续研究。该模型的Ramachandran结果[图3(a)]显示超过标准值90%以上的残基具有良好的立体化学特征(92.1%)。Verify3D结果显示超过标准值80%的残基3D-1D平均得分≥0.2(90.3%)。QMEAN[图3(b)]可以导出全局和局部绝对质量估计值。该模型的QMEAN得分为0.72,相关的Z-score为-0.03。ERRAT结果[图3(c)]显示所有残基中的76.1%的计算误差值均低于95%排斥限度。

(a)PROCHECK;(b)QMEAN;(c)ERRAT。
2.3 AA与CYP4F12活性位点的对接模式
图4(a)展示了AA的20个对接姿势的叠加图。所有的对接姿势都显示AA与Asn122和Ser399侧链形成氢键,表明其在稳定AA对接姿势中起主要作用。除上述残基外,通向活性位点的通道被大部分疏水性残基所覆盖,为非极性长链AA创造了有利的环境。对接结果的20个姿势中均显示底物与以下10个残基接触:Phe124、Ile125、Leu137、Thr324、Phe325、Phe327、Gly328、Thr332、Ile398和Ile505。
结果还显示,活性氧(Fe-O)原子与ω-2位碳原子具有距离优势,排名第一的对接姿势[图4(b)]Fe-O原子与ω-2位碳原子的距离为0.28 nm。这与上一步DFT预测的结果相一致,距离的优势更有利于在ω-2位点发生催化。

(a)20个对接结果叠加图;(b)排名第一的对接姿势(青色线为氢键)。
2.4 AA与CYP4F12动态结合过程
从对接结果中手动选择的5种不同姿势,进行500 ns的MD模拟。我们专注于分析运行之一(MD-1)。
在模拟过程中,蛋白质的构象没有剧烈变化[图 5(a)]。虽然在模拟中250 ns左右观察到蛋白质的平均RMSD值增加,但其值保持在0.4 nm范围内。在模拟过程中波动程度最大的氨基酸残基的均方根波动(RMSF)在图中相应位置标出[图5(b)],经核查其映射到蛋白质结构上高度灵活的柔性环,可以证实平均RMSD的增加是表面柔性环的正常波动所造成的而不是由于蛋白质部分的重大重组。相比之下,参与配体结合的氨基酸在模拟过程中显得稳定,并表现出很低的波动。在整个模拟过程中,AA均保持一定的稳定性,表明狭窄的结合位点仅允许受约束的构象运动[图5(a)]。

(a)CYP4F12(蓝色)和底物(红色)的时间依赖性均方根偏差(RMSD);(b)各个残基均方根波动(RMSF)。
通过3次MD模拟,收集并分析了共150 000帧轨迹中潜在取代位点到活性氧原子距离。图6是在每个MD体系下潜在代谢位点到Fe-O原子距离最小且满足距离≤0.3 nm的3D圆柱状统计图。每个体系下,ω-2位点氢原子均比其他位置的氢原子更接近Fe-O原子,活性位点残基倾向于将ω-2位点定向到Fe-O原子上。结果表明,当AA与CYP4F12活性位点结合时,ω-2位点最容易与Fe-O原子接近,这与之前结果一致。
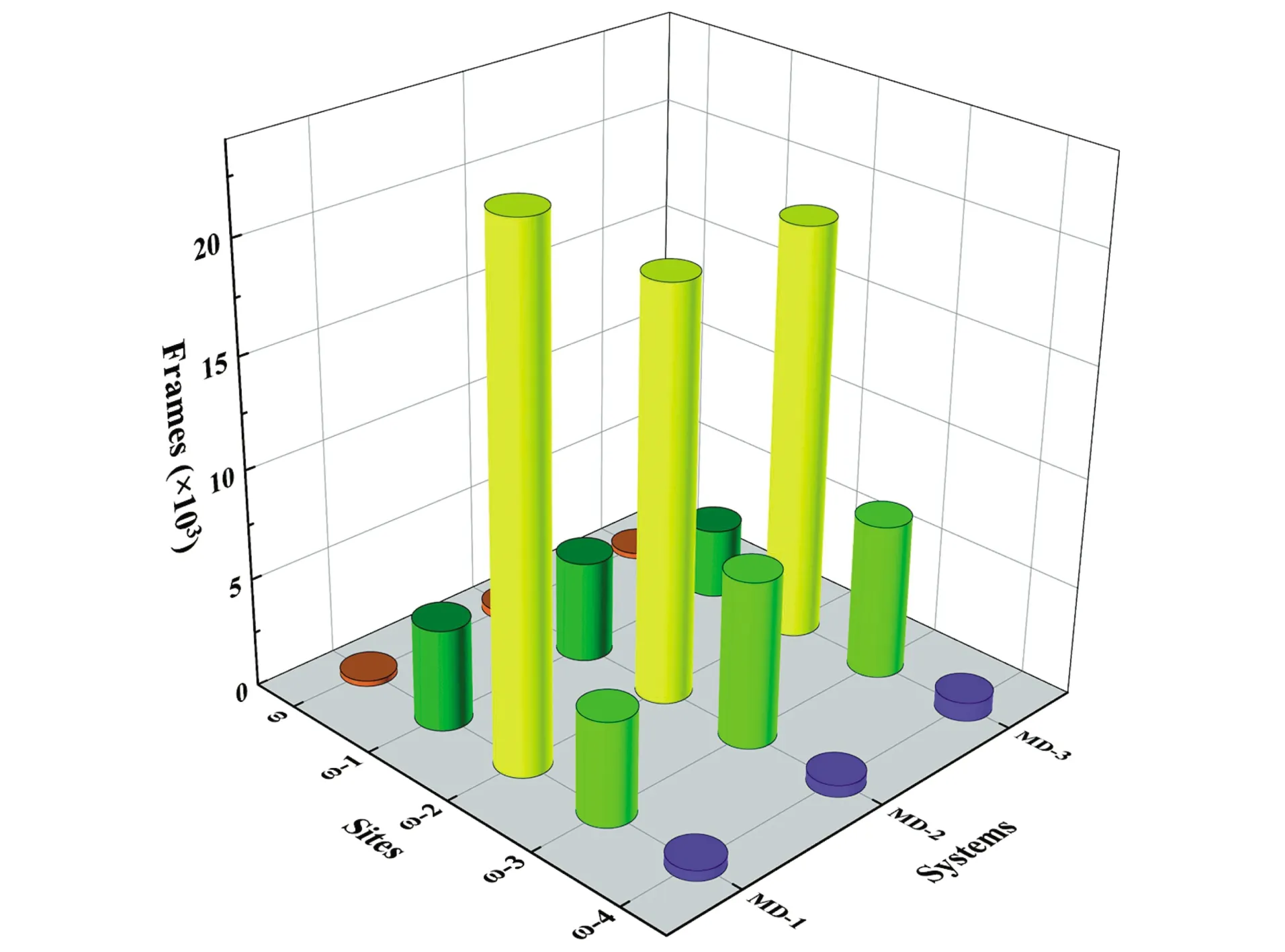
图6 在3次MD模拟中满足距离标准位点快照频率分析
我们还详细分析了CYP4F12活性位点中AA的结合模式。尽管AA的脂族链具有很高的柔性,但它在多个代表性结构中具有相似的结合模式。在500 ns模拟的最后一帧中,AA定位在血红素顶部的疏水通道内具有催化竞争优势取向。其中,ω-2位氢原子指向Fe-O原子(图7)。此外,在MD模拟期间,血红素附近的活性位点残基相对稳定,RMSF值均低于0.2 nm,且AA和残基Asn122、Ser399之间的氢键一直存在。
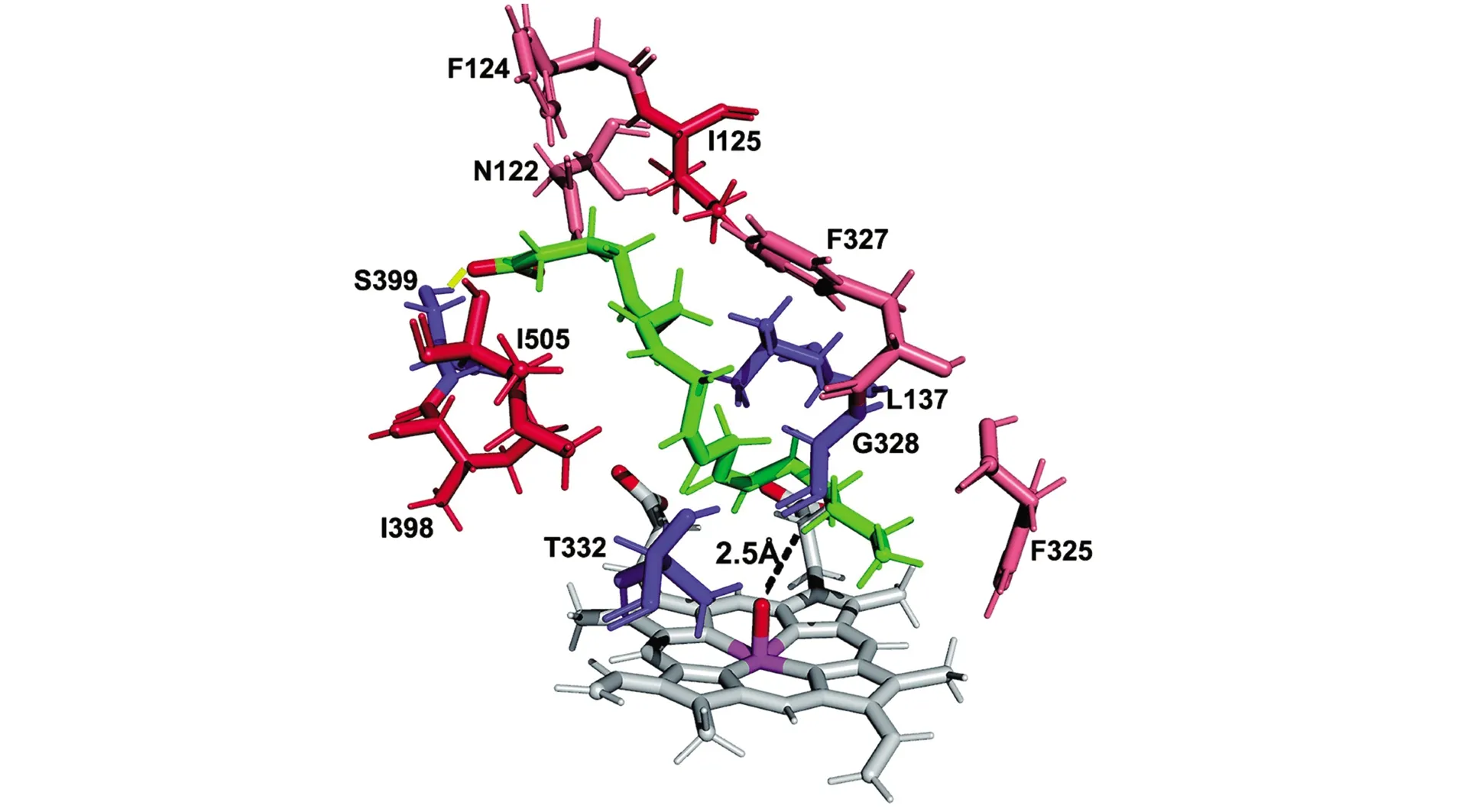
蛋白质残基的疏水性范围为红色(非常疏水)到蓝色(非常亲水)。
通过计算模拟AA与CYP4F12结合自由能得出静电相互作用是结合过程中的主要弱相互相,范德华相互作用的贡献较小。参与静电相互作用的氨基酸主要有Asn122、Ser399、Ile125、Leu137、Thr324、Gly328、Thr332、Ile398和Ile505,参与贡献范德华力的氨基酸主要有Phe327、Phe124和Phe325。
3 讨论与结论
CYP在生物体内的氧化代谢中发挥重要作用,它们催化大量不同的反应,增加母体化合物的亲水性,以促进其从人体排泄。人类对蛋白质结构与功能的研究依赖于晶体解析,而由于技术的限制,还有众多的晶体结构是未知的。同源建模无疑是一种经济、准确的方法通过预测蛋白质结构来进行前瞻性的研究。理论与实验相结合来预测化学反应各个方面的能力正在不断提高。量化计算、分子对接、分子动力学模拟等计算可以在实验观测不到的原子层面探究酶代谢的细节,精确研究结构与功能关系。
基于DFT、酶结构的同源性模型、AA与CYP4F12的分子对接和从分子对接阶段确定的实际姿势开始的500 ns MD模拟,研究AA与CYP4F12的结合。DFT计算初步氢提取反应势垒符合C—H键的提取顺序且ω-2位点具有最低的活化势垒。同源建模模型保留了CYP的整体折叠,具有12个α-螺旋和4个β-折叠,结构完整。目标蛋白与模板蛋白的几何结构对齐后的RMSD小于0.1 nm,相似度高。主要差异在一些灵活的loop环上。通过多种工具(Verify3D,ERRAT,PROCHECK和QMEAN)评估证明CYP4F12建模结构是高质量的。以上证明建模蛋白用于MD模拟是可靠的,这实际上也通过模拟的RMSD分析得到了验证。AA的pKa为4.82(http://www.drugbank.ca/drugs/DB04557),这使其在正常生理pH值下几乎没有质子化。在分子对接和动力学模拟中,AA是采用未质子化的形式。MD模拟中底物的低RMSD表明底物运动较少,其具有较低的动态性,有利于降低氧化的自由能垒。模拟过程中底物能够与Asn122、Ser399形成持续、稳定的氢键结合能力。这可以使底物很好地定位于酶活性中心,是催化发生的基础。而活性位点附近的残基Phe124、Ile125、Leu137、Thr324、Phe325、Phe327、Gly328、Thr332、Ile398和Ile505的低RMSF值也揭示了它们在催化过程中稳定底物的重要作用。在结合自由能的分析中再一次证明,以上氨基酸残基是结合过程中主要参与的氨基酸残基。
本文提出的CYP4F12与AA的结合模式,不仅为研究CYP4F12本身的活性与功能提供理论见解,而且为其他CYP4酶提供了一种研究思路与方法。与实验结果的良好匹配展现了理论作为实验伙伴的作用,以及MD模拟未来作为P450酶化学反应的可靠预测指标的作用。
