两种黄绿光铱配合物的合成及光物理性质研究
2022-06-18张筱斌窦少彬钮智刚李高楠
张筱斌,窦少彬,钮智刚,李高楠
(海南师范大学 化学与化工学院,海口 571158)
有机发光二极管(OLEDs)由于其重量轻、对比度高、视角宽、能效高以及在设计上表现出良好的通用性等优点,目前在固体照明和平板显示器领域,被认为是最有吸引力和发展潜力的技术[1-2]。重金属配合物可以同时捕获单重态和三重态激子,从而实现100%的理论内量子效率[3-4],因此对于开发有机发光二极管(OLEDs)具有至关重要的推动作用[5]。其中铱配合物具有较强的发光特性、较高的量子效率、相对较长的激发态寿命,以及良好的热稳定性、光化学稳定性等优点,因而备受人们关注,成为了OLEDs 领域内的热门研究对象[6-8]。
据大量文献报道,噻唑类铱配合物是典型橘黄色铱配合物,通过引入拉电子基能够有效的提高该类型配合物的量子产率[9-11]。基于此,本文分别以2-苯基-6-(三氟甲基)-苯并噻唑(L1)和5,7-二氟-2-苯基苯并噻唑(L2)为主配体,三苯基膦为辅助配体,合成了两种新型的铱配合物,并对它们进行了结构表征以及光物理性质测试。
1 实验
1.1 实验材料及仪器
三氯化铱(IrCl3·H2O)购自安耐吉化学。实验用2-氨基-4,6-二氟苯硫酚、2-氨基-5-(三氟甲基)苯硫醇、三苯基膦、四丁基溴化铵、苯甲酸、乙二醇乙醚、乙酸乙酯、石油醚、二氯甲烷均为市售分析纯试剂。
表征使用的仪器有核磁共振仪(NMR,Bruker AV 400 型),质谱仪(MS,Esquire HCT-Agilent 1200型),单晶衍射仪(XRD,Technologies Gemini A Ultra型),紫外分光光度计(Hitachi U-3900 型),荧光分光光度计(Hitachi F-7000 型),荧光光谱仪(Horiba FluoroMax+型)。理论计算采用Gassian09 程序[12]。
1.2 合成路线
铱配合物Ir1 和Ir2 的合成路线如图1 所示。用两种邻氨基苯硫酚和苯甲酸在亚磷酸三苯酯和四丁基溴化铵中合成配体L1 和L2;然后分别用L1和L2 两种配体在乙二醇乙醚:水(2:1)的混合溶剂中合成得到氯桥;最后氯桥与三苯基膦在二氯甲烷溶液中反应,得到两种新型铱配合物Ir1 和Ir2。

图1 铱配合物Ir1 和Ir2 的合成路线Fig. 1 Synthetic route of iridium complexes Ir1 and Ir2
1.3 合成操作
1.3.1 2-苯基-6-(三氟甲基)-苯并噻唑(L1)配体
将2-氨基-5-(三氟甲基)-苯硫醇(1.19 g,6.14 mmol)、苯甲酸(0.50 g,2.60 mmol)、四丁基溴化铵(1.32 g,4.09 mmol)和亚磷酸三苯酯(1.27 g,4.09 mmol),混合于50 mL 圆底烧瓶中,抽换气三次,在氮气保护下加热至120℃,搅拌1~2 h。冷却后加入甲醇,析出固体,抽滤,得到白色固体L1 (0.82 g,产率72.8%)。1H-NMR (400 MHz,CDCl3)δ/10-6:8.20~8.10 (m,4H),7.73 (d,J=8.8 Hz,1H),7.53~7.51 (m,3H);MS 结果为:m/z=280.0399。
1.3.2 5,7-二氟-2-苯基苯并噻唑(L2)配体
参考文献[13],采用与L1 类似的方法把2-氨基-5-(三氟甲基)-苯硫醇换成2-氨基-4,6-二氟苯硫醇,得到白色固体L2 (0.71 g,产率71.2%)。1H-NMR(400 MHz,CDCl3)δ/10-6:8.08~8.06 (m,2H),7.60~7.49 (m,4H),6.93 (dt,J1=9.2 Hz,J2=2.0 Hz,1H);m/z=248.04 [M+H]+。
1.3.3 铱配合物(Ir1)的合成
将化合物(L1) (0.30 g,1.07 mmol)和IrCl3·3H2O(0.16 g,0.54 mmol)溶解于6 mL 乙二醇乙醚与3 mL水的混合溶剂中,抽换气3 次,在氮气保护下加热到120℃,搅拌6 h。冷却后抽滤得到橙红色的铱配合物二聚体。称取二聚体(0.30 g,0.21 mmol)和亚磷酸三苯酯(0.11 g,0.41 mmol)溶解于15 mL 的二氯甲烷中,抽换气3 次,在氮气保护下,室温搅拌6 h。将所得产物通过柱层析纯化得到橙红色固体Ir1(0.12 g,产率29.2%)。1H-NMR (400 MHz,CDCl3)δ/10-6:9.63 (d,J=8.9 Hz,1H),7.85 (s,1H),7.76 (d,J=7.6 Hz,1H),7.70 (d,J=7.6 Hz,1H),7.62 (d,J=8.9 Hz,1H),7.41 (d,J=8.8 Hz,1H),7.13 (d,J=25.1 Hz,9H),7.01~6.88 (m,9H),6.76~6.67 (m,2H),6.35(d,J=7.9 Hz,1H);m/z=1011.1006 [M-Cl]+。
1.3.4 铱配合物(Ir2)的合成
用与配合物Ir1 相同的合成方法合成了配合物Ir2 (0.13 g,产率32.3%)。1H-NMR (400 MHz,CDCl3)δ/10-6:9.24~9.20 (m,1H),7.73 (d,J=7.7 Hz,1H),7.68 (dd,J=7.7,1.0 Hz,1H),7.26~7.08 (m,9H),7.08~6.87 (m,9H),6.83~6.74 (m,3H),6.72~6.67 (m,1H),6.35 (d,J=7.9 Hz,1H),5.97 (dd,J=7.5,4.7 Hz,1H);m/z=947.0887 [M-Cl]+。
2 结果与讨论
2.1 晶体结构分析
前述1H-NMR 数据和MS 数据已经确认了合成产物即为目标化合物。为进一步分析其晶体结构,将配合物Ir1 溶解在二氯甲烷和甲醇的混合溶剂中,采用溶剂挥发法得到单晶。在室温(293K)下进行了单晶XRD 测试。分析得到的晶体学数据列于表1,晶体结构如图2 所示。
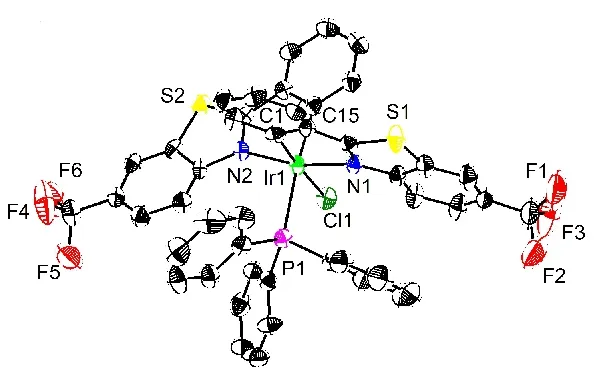
图2 配合物Ir1 的晶体结构图Fig. 2 Crystal structure of complex Ir1

表1 配合物Ir1 的晶体学数据Tab. 1 Crystallographic data for complex Ir1
根据单晶XRD 结果,该配合物是针状晶体,属于三斜晶系,空间群为P1—。Ir1 的配位环境是以铱为中心的六面体几何构型。根据键长测定结果,金属Ir 与主配体上的C 原子和N 原子的配位键长分别为Ir1-N1=0.2070(4) nm,Ir1-C1=0.2022(5) nm,Ir1-N2=0.2077(3) nm,Ir1-C15=0.2055(4) nm;与辅助配体上的P 原子的配位键长为Ir1-P1=0.2455(11)nm;与Cl 原子的配位键长为Ir1-C11=0.2491(11)nm。由此可见,Ir 与主配体上原子的配位键键长要比与辅助配体以及Cl 原子的短。根据键角测定计算结果可知,该配合物的主要键角中C15-Ir1-P1 和C1-Ir1-Cl1 分别为175.25(14)°和176.00(11)°,近180°。其它的键角,∠N1-Ir1-Cl1=99.21(11)°、∠N1-Ir1-P1=88.87(9)°、∠C1-Ir1-P1=94.82(10)°、∠C1-Ir1-N2=92.63(17)°、∠N2-Ir1-Cl1=87.00(10)°、∠C1-Ir1-C15=89.22(16)°、∠C15-Ir1-Cl1=86.79(13)°、∠C15-Ir1-N1=89.35(16)°,都接近于直角90°,而且所有键角与报道的类似Ir 配合物相近[14]。结构分析得到的Ir1 晶体学数据已收录在剑桥晶体学数据中心(No.CCDC 2083030)。
2.2 光谱学表征
铱配合物Ir1 和Ir2 常温下在二氯甲烷溶液中的紫外吸收光谱和荧光发射光谱如图3 所示,相应的光物理数据列于表2。由图3 可见,两个化合物的吸收光谱和发射光谱谱图十分相似。对于紫外吸收光谱(图3(a)),小于350 nm 的吸收带归属于配体为中心的π-π*跃迁;350~500 nm 之间的弱吸收带归属于金属-配体的1MLCT 跃迁和配体内部的1ILCT跃迁。在荧光发射光谱(图3(b))中,Ir1 的最大发射波长是548 nm,Ir2 的发射波长是546 nm,两种配合物发光颜色都属于黄绿色发光。经测试,以fac-Ir(ppy)3作为参考标准(Φem=0.4)[15],Ir1 和Ir2的相对量子产率分别是41.1%和69.2%,并且引入了2 个F 原子的配合物Ir2 的量子产率有显著提高。这可能是因为C-F 键不仅可以有效降低非辐射失活速率进而提高材料的发光效率,而且还可以在空间上改变分子堆积并降低自猝灭行为[16]。

表2 配合物Ir1 和Ir2 的光物理数据Tab. 2 Photophysical data of complexes Ir1 and Ir2

图3 铱配合物Ir1 和Ir2 在二氯甲烷溶液中的紫外吸收光谱(a)和荧光发射光谱(b)Fig. 3 UV-Vis (a) and FL (b) spectra of iridium complexes Ir1 and Ir2 in CH2Cl2 solution
2.3 理论计算
采用Gassian09 程序[12]对配合物进行了理论计算研究,使用了B3LYP 方法在6-31G 基组上对它们进行了结构优化,随后利用密度泛函理论(DFT)计算了它们的紫外吸收和荧光发射性质。计算的分子前线轨道(HOMO 和LUMO)如图4 所示,计算的自旋允许的电子跃迁列于表3。由图4 可知,配合物Ir1 和Ir2 的HOMO 轨道都主要分布在金属Ir和C^N 主配体上,LUMO 轨道主要分布在环金属C^N 主配体上。计算结果表明,Ir1 和Ir2 的紫外光谱中最低能量跃迁来自HOMO→LUMO (表3 中S1),因此可归因于MLCT 和ILCT 跃迁,进一步证实了紫外吸收光谱的实验结果。为了深入研究这些配合物的荧光发射光谱,计算了最低三重态激发态,它们的跃迁类型同样为HOMO→LUMO (表3 中T1)。这些结果证实了Ir1 和Ir2 的激发态为金属-配体/配体内部的电荷转移(3MLCT/3ILCT),与荧光光谱中观察到的振动结构一致。
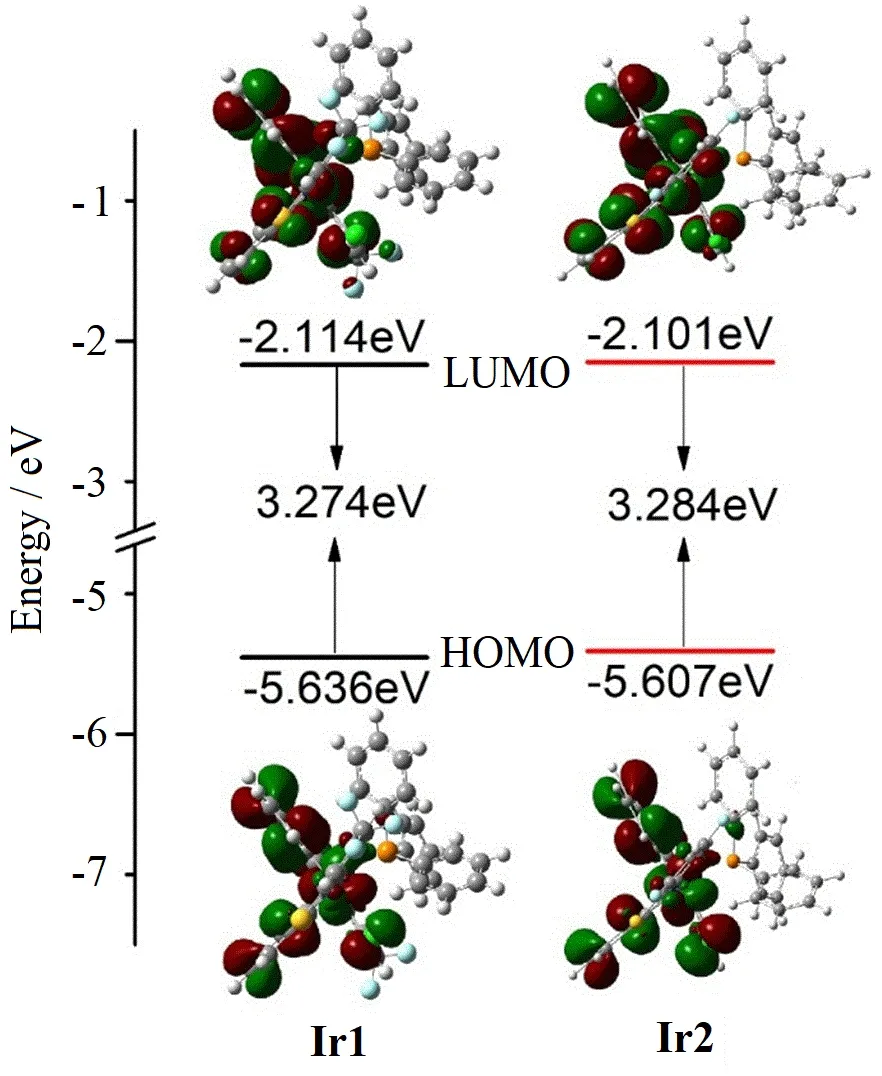
图4 DFT 计算的配合物Ir1 和Ir2 的前线分子轨道能级图Fig. 4 The frontier molecular orbital energy level diagram of complexes Ir1 and Ir2 by DFT calculation

表3 配合物Ir1 和Ir2 最低单重态和三重激发态的主要电子跃迁贡献和特征Tab. 3 Main contributions and transition characters the lowest singlet and triplet excited states of complexes Ir1 and Ir2
3 结论
1) 以氟取代的氨基苯硫酚和苯甲酸为原料合成出两种氟取代的苯并噻唑类主配体,再与三氯化铱合成相应的氯桥,最后通过与三苯基膦配位合成出配合物Ir1 和Ir2,产率约30%。1H-NMR 和MS表征结果表明合成产物为目标化合物,配合物Ir1的分子结构经单晶XRD 分析得到确认。
2) 紫外可见吸收光谱和荧光发射光谱测试表明2 种配合物光谱性质接近,Ir1 和Ir2 的最大发射光谱波长分别为548 和546 nm,均为发黄绿光的铱配合物,相对量子产率分别为41.1%和69.2%,荧光寿命分别为0.65 和0.66 μs。密度泛函理论(DFT)计算结果表明,紫外最大吸收峰和荧光发射峰均归因于MLCT/ILCT 跃迁。
