咪唑/三唑羧酸配体配合物的合成及性能研究
2022-06-17刘博予岳天才张开发王多志
刘博予, 岳天才, 张开发, 王多志
(新疆大学 化学学院 省部共建碳基能源资源化学与利用国家重点实验室,新疆 乌鲁木齐 830017)
有机-金属配合物是一类具有永久孔隙率和超高表面积的无机—有机杂化材料[1-2],凭借高比表面积、可调节孔径和有机基团修饰的特性,在材料科学中享有独特地位,在学术和工业环境研究领域有诸多应用,例如气体储存和分离[3-4]、催化[5]、荧光[6]、传感[7-8]、光学和磁性材料[9-11]等。在已经报道的有机-金属配合物中,其总体设计可分为两部分,结合域(金属簇)和结构域(骨架)。不同金属离子簇与多样的有机骨架结合能够形成新的拓扑结构,从而得到具有功能性的多孔材料,进一步丰富了金属有机配合物领域的研究和应用。另外有机-金属配合物的孔道为底物分子提供宿主空间,在空间尺寸效应下,底物分子和有机-金属配合物材料之间存在强烈相互作用。由于羧酸类配体配位键的稳定性和有机配体的可设计性,成为合成有机-金属配合物的主流。当羧基与高价金属进行构建时,可以形成强配位键[12],且羧酸根具有负电荷,有利于提高孔洞率和稳定性,合成的配合物具有多种配位模式,从而形成理想的网络结构。咪唑/三唑类配体中具有sp2杂化的氮原子,当其和羧酸根进行组合后,使配体同时具备羧酸根和多氮类配体的优点,从而大大增加所得有机-金属配合物的稳定性。羧酸配体在构建分子磁体方面同样存在着巨大的潜力,这些分子磁体具有反铁磁性、铁磁性和自旋旋转等性质[13-15]。含杂环咪唑/三唑基的有机配体是另一种基于簇的桥接配体,有机-金属配合物显示自旋禁阻,反铁磁性,长程磁性有序,变磁性和自旋倾斜[16-18]。因此,羧酸咪唑/三唑基团与金属离子结合形成有机-金属配合物可能会产生多样的结构和有趣的性能[19-20]。
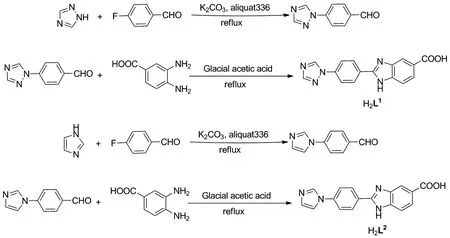
本文合成了咪唑/三唑基的两种羧酸配体H2L1和H2L2(H2L1=2-(4-(1H-1,2,4-三唑-1-基)苯基)-1H-苯并[d]咪唑-5-羧酸;H2L2=2-(4-(1H-咪唑-1-基)苯基)-1H-苯并[d]咪唑-5-羧酸)(Scheme 1),在溶剂热条件下合成3三种新的配合物[Co(HL1)2(H2O)4](1),[Ni(HL1)2(H2O)4](2)和[Mn(HL2)2(H2O)4](3),通过IR和X-射线单晶衍射对配合物结构进行表征,结果表明配合物1~3具有相似的晶体结构,通过分子间氢键构筑成二维超分子结构,并对配合物1~3的磁性和电化学性能进行了研究。
1 实验部分
1.1 仪器与试剂
INO-VA-400 MHz型核磁共振仪(DMSO-d6为溶剂,TMS为内标);Brucker Equinox55FT-IR型红外光谱仪(KBr压片);Bruker APEX II Smart CCD型X-射线单晶衍射仪;Bruker D8型X-射线粉末衍射仪;DSC200F3型差示扫描量热析仪;MPMS-XL-7型超导量子干涉磁强计;VMP-300型电化学工作站。
所用的试剂均为分析纯,除特别说明外,未经处理直接使用。
1.2 合成
(1)H2L1和 H2L2的合成(以H2L2为例)
4-(1H-咪唑-1-基)苯甲醛的合成:将3.40 g(50 mmol)咪唑,5.0 g(36 mmol)K2CO3加入到100 mL 的圆底烧瓶中,加入五滴相转移催化剂甲基三辛基氯化铵,再加入30 mL DMF,升温至90 ℃,反应5 min后,将对氟苯甲醛6.2 g(50 mmol)分三次滴加到反应混合物中,滴加完毕,将温度调节至80 ℃,反应10 h。冷却至室温,将冷却的反应混合物倒入150 mL的冰水中搅拌,有大量黄色沉淀析出,冰箱中冷冻30 min,使得反应物析出完全,抽滤,干燥,得到黄色固体,产率为91%[21]。
H2L2的合成:将0.86 g(5 mmol)4-(1H-咪唑-1-基)苯甲醛和0.76 g(5 mmol)3,4-二氨基苯甲酸,加入到25 mL的圆底烧瓶中,加入15 mL冰醋酸。混合物加热回流12 h,然后冷却至室温,将反应混合物倒入50 mL冰水中,搅拌,抽滤,用蒸馏水洗涤得到淡绿色固体H2L2,产率为65%[22](合成步骤见Scheme 1)。
Scheme 1
用1,2,4-三氮唑代替咪唑,用相似的方法合成H2L1。
H2L1:灰色固体,收率73%;1H NMRδ: 9.42(d,J=9.3 Hz, 1H, 1,2,4-triazole), 8.37(d,J=8.8 Hz, 2H, 1,2,4-triazole), 8.31(s, 1H, benzimidazole), 8.21(s, 1H, benzimidazole), 8.12(d,J=8.8Hz, 2H, benzene), 7.88(dd,J=8.5 Hz, 1.5 Hz, 1H, benzene), 7.70(d,J=8.5Hz, 1H, benzene); IRν: 3298(O—H), 3123(N—H), 1705(C=O), 886(C—H), 844(C—H)cm-1。
(2)配合物1~3的合成(以配合物1为例)
将Co(ClO4)2·6H2O(36.6 mg, 0.1 mmol),H2L1(30.5 mg, 0.1 mmol)加入到20 mL的聚四氟乙烯反应釜中,加入2 mL H2O和4 mL CH3CN,以1.3 ℃/min的速度升温至160 ℃,反应12 h。冷却至室温得黄褐色配合物单晶1。
用NiCl2·6H2O代替Co(ClO4)2·6H2O,用类似方法合成配合物2。
用MnCl2·4H2O代替Co(ClO4)2·6H2O,将配体变为H2L2,用类似方法合成配合物3。
1: 产率约52%(以Co计);IRν: 3440, 3133, 1947, 1895, 1664, 1597, 1520, 1495, 1478, 1454, 1438, 1417, 1367, 1319 , 1257, 1243, 1216, 1144, 1127, 1084, 1050, 966, 896, 854, 786, 727, 701, 669, 647, 561, 491, 462, 440 cm-1。
2: 产率约43%(以Ni计);IRν: 3367, 3897, 3850, 3833, 3796, 3134, 2401, 1947, 1895, 1665, 1597, 1521, 1478, 1455, 1438, 1418, 1367, 1319, 1277, 1243, 1217, 1144, 1127, 1084, 1051, 1017, 965, 896, 854, 786, 727, 700, 669, 646, 560, 491, 463, 440 cm-1。
3: 产率约67%(以Mn计);IRν: 3373, 3189, 3132, 2399, 1917, 1808, 1668, 1621, 1596, 1543, 1508, 1478, 1446, 1425, 1368, 1302, 1256, 1126, 1084, 1063, 1015, 964, 948, 907, 840, 788, 730, 680, 649, 564, 487, 439 cm-1。
1.3 晶体结构测定
在常温或者低温条件下,采用型号为Bruker Smart Apex-Ⅱ CCD衍射仪对配合物1~3进行晶胞的测试,晶胞参数的收集是用石墨单色铜靶和钼靶的X射线,用SADABS程序[23]进行数据的经验校正吸收,用SAINT程序[24]进行数据的还原。用直接法解析出晶体的结构,用SHELXTL-97程序[25]通过全矩阵最小二乘法进行数据的精修。用衍射角的各向异性确定非氢原子,确定氢原子是通过几何计算和电荷守恒进行理论加氢的方法。部分存在乱序的配合物依据合适的占据位点来分离进行精修。配合物1~3的CCDC号分别为2016867、2016868、2016869,配合物1~3的晶体学参数列于表1,重要的键长、键角列于表2。

表1 配合物1~3的晶体学参数

表2 配合物1~3的主要键长和键角
2 结果与讨论
2.1 晶体结构
晶体结构解析表明,配合物1和2具有相似的晶体结构,因此只详细讨论1的结构。配合物1属于单斜晶系,C12/c1的空间群,单核零维结构,在氢键的作用下,构筑成二维超分子结构。不对称单元包含一个晶体学独立的Co(Ⅱ)离子、两个H2L1-1阴离子和四个水分子,如图1a所示。六配位Co(Ⅱ)离子与两个氮原子和四个氧原子连接,形成八面体配位构型,其中两个氮原子(N1)来自两个H2L1-1配体,四个氧原子(O1W和O2W)来自四个配位水分子,金属Co(Ⅱ)离子占据八面体配位构型的中心,两个氮原子占据配位构型的轴向位置,四个氧原子占据配位构型的赤道位置。两个配合物的N1#1—Co1—N1和N1#1—Ni1—N1的角度都为180°。而配合物1的Co—O键键长范围在2.081(3)~2.115(3)Å之间,Co—N键键长为2.147(3)Å。配合物2的Ni—O键键长范围在2.059(2)~2.077(2)Å之间,Ni—N键键长为2.091(3)Å。这个单核的结构单元通过配位水分子的氧原子和氢原子与H2L1配体羧酸基团的氧原子形成氢键(O2W—H2WB…O1#2,键长1.70Å,键角163.3°,对称代码:#2-x+1,-y+1,-z+1)进而形成一维链状结构,如图1b所示。进一步通过氢键(O2W—H2WA…O1#5,键长1.85Å,键角148.0°,对称代码:#5 x+1,-y+1, z+1/2)将一维链连接形成二维平面结构,如图1c所示。
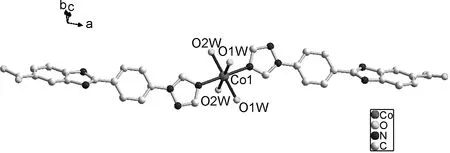
(a)
晶体结构解析表明,配合物3属于单斜晶系,C12/c1的空间群,单核零维结构,在氢键的作用下,构筑成二维超分子结构。不对称单元包含一个晶体学独立的 Mn(Ⅱ)离子、两个H2L2-1阴离子和四个配位水分子,如图2a所示。六配位Mn(Ⅱ)离子与两个氮原子和四个氧原子连接,形成八面体配位构型,其中两个氮原子(N1)来自两个H2L2-1配体,四个氧原子(O1W和O2W)来自于4个水分子,金属Mn(Ⅱ)离子占据八面体配位构型的中心,两个氮原子占据配位构型的轴向位置,四个氧原子占据配位构型的赤道位置。配合物3的N1#1—Mn1—N1角度为180°, Mn—O键键长范围2.1803(16)~2.1884(16)Å之间,Mn—N键键长为2.2419(19)Å。这个单核的结构单元通过配位水分子的氧原子和氢原子与H2L2配体羧酸基团的氧原子形成氢键(O1—H1B…O3#2,键长1.92Å,键角173.7°,对称代码:#2-1+X, +Y, +Z)和(O2—H2A…O4#3,键长1.83Å,键角166.3°,对称代码:#3 1-X, 1-Y, 1-Z),且沿着a轴方向有序排列进而形成一维链状结构,如图2b。最后通过氢键(O2—H2B…O4#3,键长1.96 Å,键角145.1°,对称代码:#4-1+X, 1-Y,-1/2+Z)将一维链沿着b轴方向连接形成二维平面结构,如图2c。

(a)
2.2 X-射线粉末衍射(PXRD)分析
利用X-射线粉末衍射(PXRD)技术,确定配合物1~3相纯度,如图3a~3c。通过对测试数据与模拟数据对比分析显示,实验所得的数据曲线与X-射线单晶衍射数据的模拟曲线非常吻合,这表明1~3具有良好的相纯度,只有峰值强度存在差异,可能由于数据收集过程中微晶的非随机取向所导致。
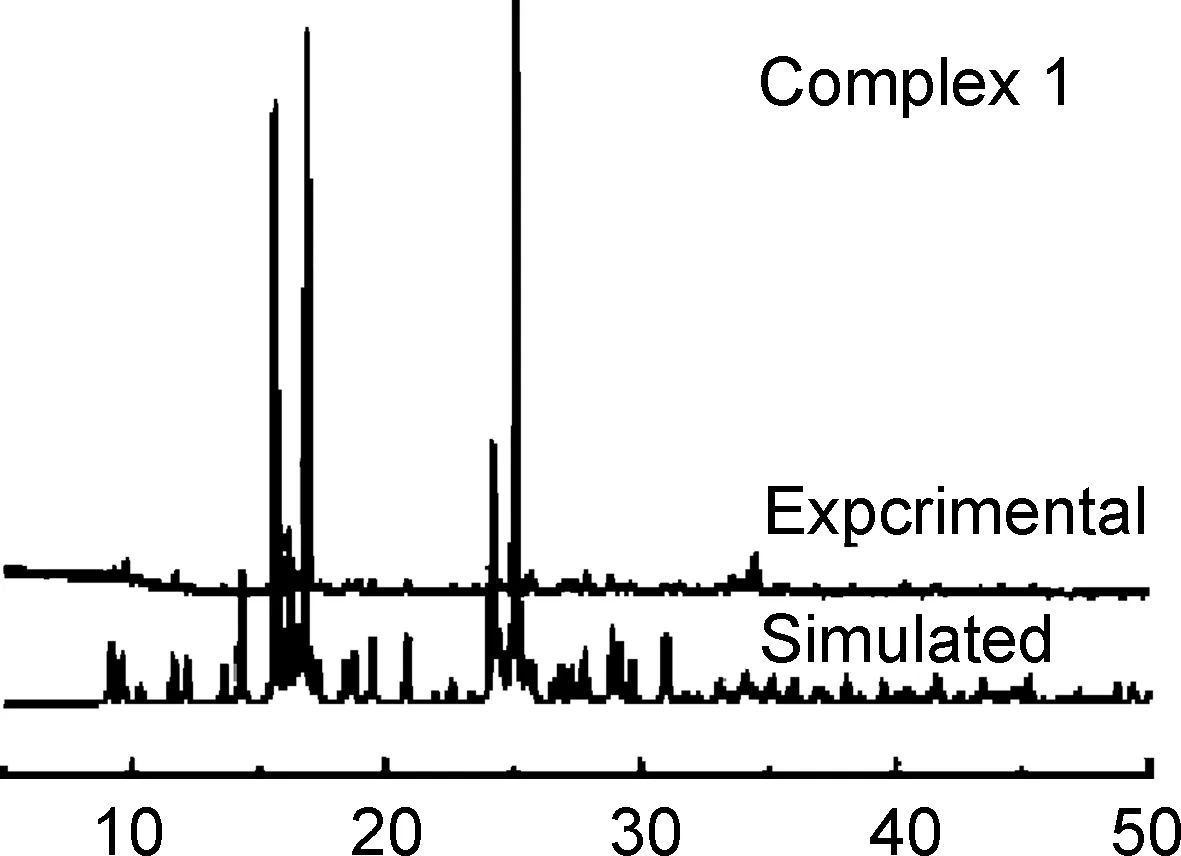
2θ/(°)(a)2θ/(°)(b)2θ/(°)(c)
2.3 热重(TG)分析
热稳定性是评价配合物性能的重要因素之一,在氮气条件下及25~900 ℃的温度区间内,用热重分析法测定了配合物1~3的热稳定性,如图4所示。配合物1~3存在两个明显的失重阶段,配合物1的第一个阶段在175~310 ℃的温度区间,失重比例:测试值为9.48%,理论值为9.51%,配合物2的第一个阶段在210~330 ℃的温度区间,失重比例:测试值为8.55%,理论值为9.52%,配合物3的第一个阶段在172~350 ℃的温度区间,失重比例:测试值为8.71%,理论值为9.59%,由晶体结构中配位水分子的挥发所致。配合物1~3的第二个阶段分别从310 ℃、330 ℃和350 ℃开始,重量急剧下降,是由高温导致的晶体框架坍塌导致。而配合物分解的剩余物一般为金属氧化物。
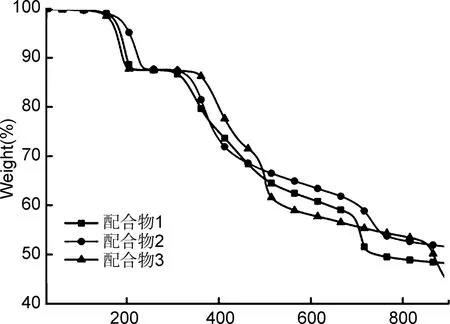
Temperatur/℃
2.4 磁性
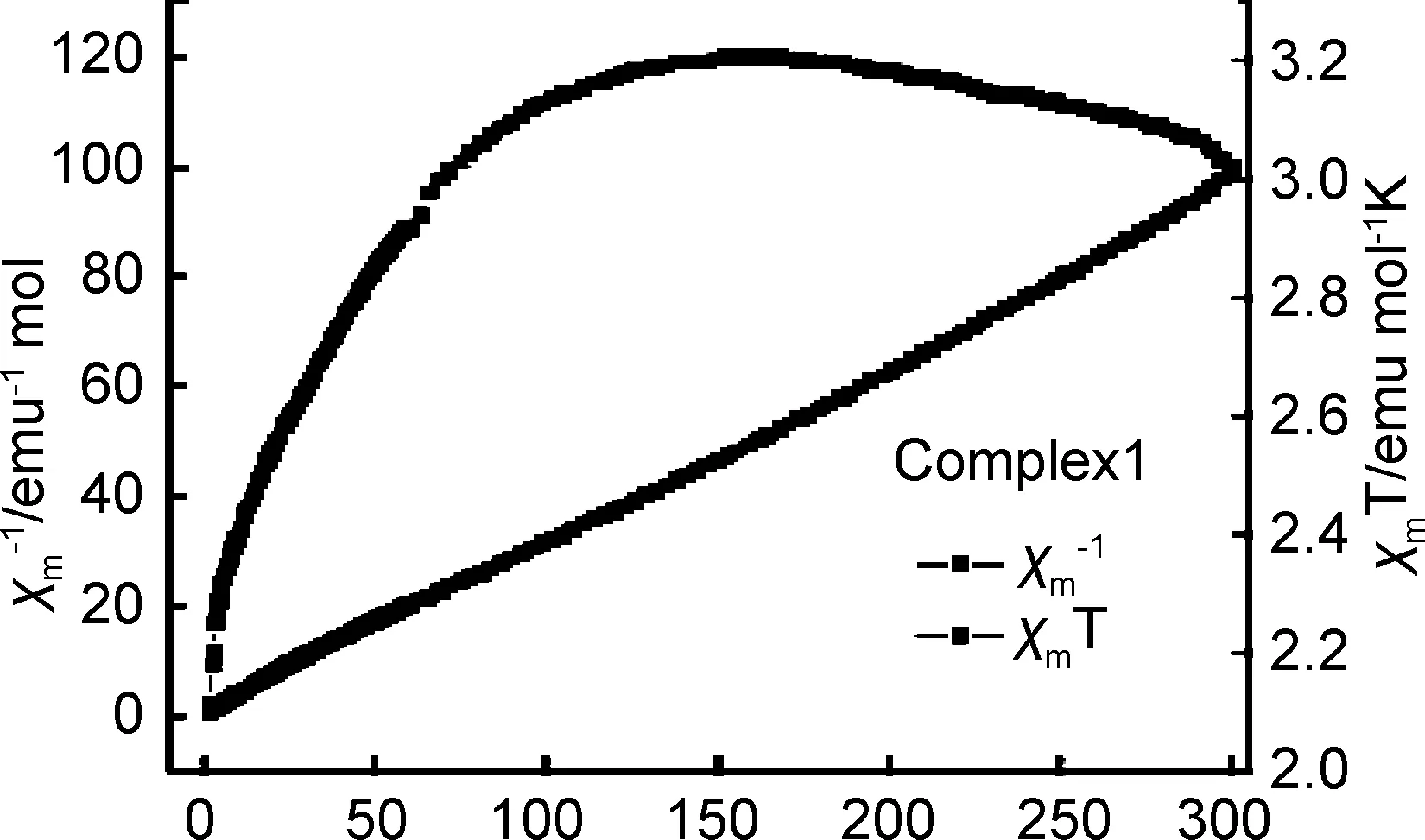
对配合物1~3的磁性进行了研究,测试所加外源磁场的强度为1000 Oe,实验所需温度为2~300 K,配合物1~3的χm-1和χmT随着温度T变化的曲线图,如图5a~5c所示。在300 K时配合物1的χmT值为3.02 emu mol-1K,低于磁学上孤立的高自旋的Co(Ⅱ)离子,自旋值为3.75 emu mol-1K(g=2.0)。在逐渐冷却过程中,χmT值先逐渐升高,在155 K时达到最大值3.21 emu mol-1K,之后迅速下降,到2 K时,χmT的值达到最小,为2.11emu mol-1K,通过曲线的变化趋势以及Curie-Weiss定律计算得到值C=3.17 emu mol-1K和θ=3.22 K,以此确定配合物1具有顺磁性相互作用,如图5a。在300 K时配合物2的χmT值为0.65 emu mol-1K,低于磁学上孤立的高自旋的Ni(Ⅱ)离子,自旋值为1.17 emu mol-1K(g=1.1)。在逐渐冷却过程中,χmT值先逐渐升高,在17 K时达到最大值1.32 emu mol-1K,之后迅速下降,到2 K时,χmT的值达0.76 emu mol-1K,通过曲线的变化趋势以及Curie-Weiss定律计算得到值C=0.75 emu mol-1K和θ=-19.12 K,以此确定配合物2为反铁磁性。在300 K时配合物3的χmT值为4.4 emu mol-1K,略高于磁学上孤立的高自旋的Mn(Ⅱ)离子,自旋值为4.38 emu mol-1K(g=2.0)。在逐渐冷却过程中,χmT值先逐渐升高,在83 K时达到最大值4.57 emu mol-1K,之后迅速下降,到2 K时,χmT的值达到最小,为4.29 emu mol-1K,通过曲线的变化趋势以及Curie-Weiss定律计算得到值C=4.35 emu mol-1K和θ=-6.16 K,以此确定配合物3为反铁磁性。
T/K(a)
2.5 电化学性能
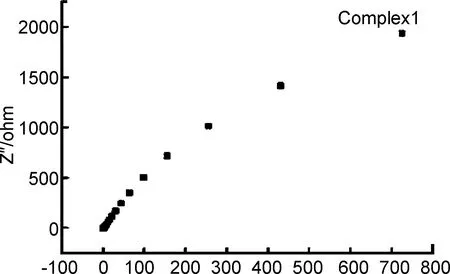
在室温条件下,对配合物1和3的电化学性能进行测试,如下图。在不同电压下,配合物1和3的循环伏安曲线图如图6所示,由图可知,配合物1和3的所有氧化还原峰随着电压强度的递增峰强度逐渐增大。由此可知,配合物1和3中的金属离子在电极表面发生了电荷的转移,工作电极中的活性物质在电极表面发生了氧化态和还原态之间的相互转化,使得电子发生移动形成了电流。同时在不同电流强度下,充放电时均出现平台期,这个平台期的电压正好与循环伏安谱图中的氧化峰和还原峰相对应,从侧面说明充放电的平台可能是由于材料的氧化还原导致。充放电曲线如图7所示,表明随着电流密度的增大充电和放电的时间大大缩短,根据公式C=It/(mΔV)可以求出1和3对应电流密度条件下的电容量,由此可计算得到随电流密度的增大1和3电容量依次为83.61 , 76.03 , 68.92 , 60.63 , 48.79 Fg-1和36.35 , 32.88 , 27.69 , 23.88 , 16.62 Fg-1,由递减的数值可以看出每进行一次充放电其电容量都会有损失。最后对其交流阻抗进行了测试如图8所示,由曲线显示可知,曲线下端的半圆形状的半径代表这种物质自身的电阻,曲线上端近似倾斜直线部分代表活性物质与电解液之间的电阻,由此可知这样的功能材料可以作为电容器的电极材料,其导电性受两部分的电阻影响。

Potential(V vs.SCE)Potential(V vs.SCE)

Time/s
Z′/ohm(a)
以咪唑/三氮唑两种羧酸为配体,与钴、镍、锰等三种金属盐在溶剂热条件下合成三种新的配合物。晶体结构分析表明,配合物1~3具有相似的晶体结构,属于单核零维结构,金属中心离子均为六配位的八面体配位构型,在氢键作用下构筑成二维超分子结构,不同的是咪唑以及三氮唑中的氮原子与金属发生配位,且氢键的位置存在差异。对配合物1~3进行磁性性质的研究,发现配合物1具有顺磁性相互作用,配合物2和3具有反铁磁性相互作用。并对1和3进行电化学性能测试,结果表明两种配合物可以作为潜在的电容器电极材料。
