质子交换膜电解制氢氢气渗透研究进展
2022-06-16郭志远徐桂芝邓占锋李宝让
叶 青,宋 洁,侯 坤,郭志远,徐桂芝,邓占锋✉,李宝让
1) 国网智能电网研究院有限公司先进输电技术国家重点实验室,北京 102209 2) 华北电力大学能源动力与机械工程学院,北京 102206
氢气具有能量密度高、无污染、可长时间存储等优势,能够作为连接不同能源形式的桥梁,是跨能源网络的理想互联媒介.氢能产业包含制−储−用等关键环节[1−2],其中,制氢作为基础,对于整个产业发展至关重要.质子交换膜(PEM)电解制氢,作为适应可再生能源制氢的先进技术,是国内外大力发展的热点[3].不但可以促进新能源规模化消纳,绿电制取的氢气还可以广泛用于交通运输、合成氨、氢冶金等,实现多领域深度碳减排[4−5].基于PEM优异的机械性能与气体隔绝能力,高压PEM电解堆已实现商业化[6−8].但由于膜的吸水特性,在高压PEM电解堆运行过程中,仍存在气体渗透问题[9−10].在低电流密度区,气体渗透与安全、效率密切相关,而在高电流密度区,渗透影响膜的衰减[11−13].因此,明晰气体渗透的理论机制,研究不同运行参数对渗透的影响规律,对于质子交换膜电解堆的安全、高效运行至关重要.
目前,在质子交换膜电解制氢气体渗透的相关研究中,一方面采用理论计算方法,通过建立渗透模型,对渗透率进行定量计算;另一方面利用试验测量方法,测试环境包括渗透池与电解池两类.二者区别在于渗透池可以模拟电解制氢运行时的温度、压力等条件,但无法施加电流,而在电解池中进行原位测试时,能够全面评估各类参数对渗透的影响,特别是不同运行电流密度下的气体渗透行为.此外,在质子交换膜电解制氢环境中,相对于氧气,氢气渗透产生的影响更为严重,本文主要综述了各类操作条件变化对氢气渗透影响规律的研究进展.
1 渗透基本理论
粒子或分子随机热运动引起的布朗运动将导致扩散,分子在距离d上的浓度差Δc会产生渗透通量Φ,如菲克定律所描述:

其中,D表示分子在介质中的扩散系数.
根据亨利定律,介质中溶解气体的浓度cgas与其分压pgas及在介质中的溶解度Sgas密切相关,可以表示为:

由扩散引起的气体渗透率εgas是扩散系数与该气体在介质中溶解度的乘积:

当膜将两个分压不同的腔室隔开时,利用式(2)与(3),菲克定律可以表示为分压差Δpgas的函数[14]:
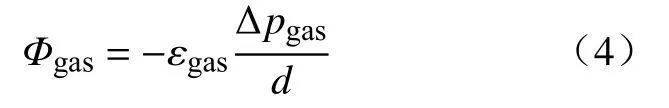
由式(4)可以看出,影响气体渗透的因素主要有以下几个方面,一是温度、压力等外界参数,通过影响扩散系数Dgas与溶解度Sgas对渗透率产生影响;二是水合程度、厚度等膜的特性;三是膜两侧的分压差.此外,在电解制氢实际工况中还存在运行电流,下面将从上述几方面对氢气渗透的影响规律进行综述.
2 温度/压力对氢气渗透率的影响
Ito等[15],Battino和Clever[16],以及 Mann等[17]总结了氢气在水中的溶解度S随外界温度与压力的变化规律,如图1所示.可以看出,当压力处于0.1~10 MPa范围内,溶解度保持相对稳定,而当压力高于10 MPa时,溶解度随压力的升高而降低.基于此,Battino等[16]提出了0.1 MPa分压、273~353 K条件下氢气在水中的溶解度公式:

图1 氢气在液态水中溶解度S随温度T变化的阿瑞尼斯图[15-17]Fig.1 Arrhenius plots of hydrogen solubility ( SH2) in liquid water[15-17]

其中,SH2为氢气在水 中的溶解度,mol·m−3·Pa−1;T为温度,K.
Wise和Houghton[18]测试了氢气在 10~60 °C范围内的扩散系数,提出了扩散系数与温度的函数关系式,其中D0=(4.9±0.3) cm2·s−1,ED=(16.51±0.17) kJ·mol−1:
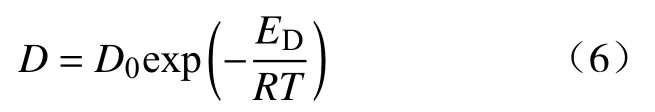
其中,D为扩散系数,cm2·s−1;ED为扩散激活能,J·mol−1;R=8.314 J·mol−1·K−1,为气体常数;T为温度,K.
在高压PEM电解制氢的常规运行压力范围(3.5 MPa)内,扩散系数与溶解度主要受温度影响,而压力产生的影响很小.因此,渗透率也主要与温度相关.图2为氢气在不同干湿状态Nafion117膜中渗透率与温度关系的Arrhenius曲线[15,19−26].可以看出,随温度升高,渗透率呈增大的趋势.Nafion湿膜中的氢气渗透率约为干膜的5~10倍,Nafion干膜与聚四氟乙烯(PTFE)的氢气渗透率接近.

图2 氢气在不同干湿状态下Nafion117质子交换膜中渗透率的阿瑞尼斯图[15, 19−26]Fig.2 Arrhenius plots of hydrogen permeability in a Nafion 117 membrane at dry and wet conditions[15, 19-26]
3 膜水合程度对氢气渗透率的影响
Schalenbach等[14]通过试验测量了膜在不同水合程度下的氢气渗透率,测量是在80 °C、膜两侧压力为0.1 MPa(分别为H2与N2)条件下进行的,通过对膜吹扫不同相对湿度的气体,使Nafion 212(NR 212)膜的水合程度发生变化,结果如图3所示.可以看出,氢渗透率随相对湿度的升高而增加.膜的水合程度随气体相对湿度的增加而提高,当膜发生水合时,水以水通道的形式积聚,由于水的氢气渗透率是干膜的5~10倍,通过水通道的气体渗透可以绕过固相路径(图4),从而增大了氢渗透率.
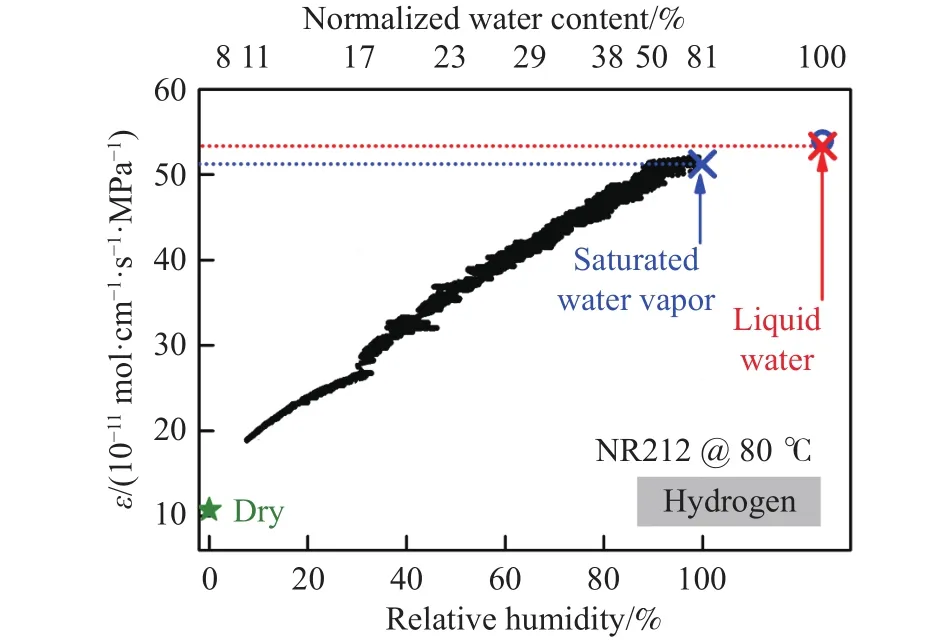
图3 Nafion212膜在80 °C条件下的渗透率与相对湿度、归一化水含量的关系[14]Fig.3 Permeability of Nafion 212 at 80 ℃ as a function of relative humidity and normalized water content[14]
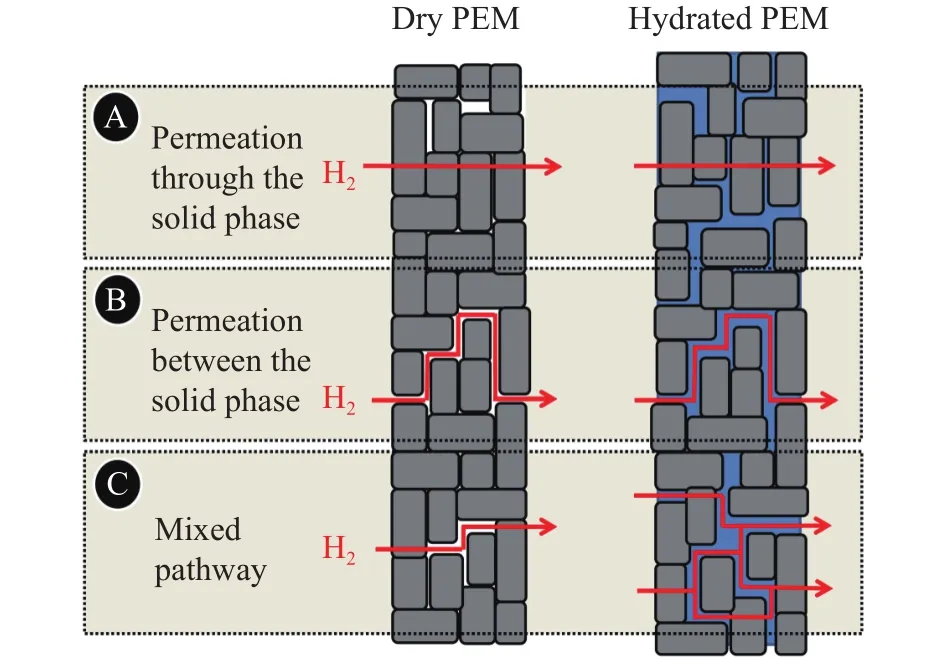
图4 氢气以不同路径渗透通过PEM的示意图(灰色区域代表固相,蓝色区域代表水相, 白色代表孔洞; 左侧为干膜, 右侧为湿膜)[14]Fig.4 Descriptive sketch of the pathways for gas permeation through a segment of PEM exemplified for hydrogen molecules (The solid polymeric phase is depicted as the gray area, water as the blue area, and pores filled with gas as the white area; left: dry PEM; right: hydrated PEM)[14]
为了进一步解释氢气渗透率随Nafion膜水合程度的变化关系,并对气体在水合膜中的渗透机制进行分析,基于水合Nafion膜的微观结构,Schalenbach等[27]建立了渗透模型并开展理论计算.通过在水相与固相之外加入中间相(图5),并改变中间相与固相的渗透率,将模型确定的总渗透率与实测数据进行拟合.结果发现,只有放大中间相与固相的渗透率时,模拟数据才能与实验值一致.与仅通过水相的模拟渗透率相比,通过所有三个相的交替渗透使整体渗透率增加了5.4倍,即水合膜约81%的氢渗透率归因于中间相和固相.该研究对水合Nafion膜中氢气渗透的路径提出了新的观点,作者进一步推断了中间相与固相渗透率增大的原因,中间相较高的氢气渗透率源于水相中的氢键网络不太明显,且在该状态下范德华力较弱,而固相渗透率的增加源于聚合物基质因吸水而软化,因为水可充当聚合物基质的增塑剂.

图5 Nafion膜三维结构模型的截面图,图中横纵坐标表示200个网格单位,每个网格单位对应边长为0.21 nm的立方体(灰色区域: 固相,蓝色区域: 水相, 绿色区域: 中间相)[27]Fig.5 Two-dimensional cross section of the modeled three-dimensional cubic structure of Nafion with an edge length of 200 segments.One segment of the mesh corresponds to a cube with 0.21 nm edge length.(Gray area: solid phase; blue area: aqueous phase; green area:intermediate phase)[27]
4 压差的影响
4.1 渗透与氢气分压差的线性关系
Schalenbach等[14]研究了Nafion117膜的氢渗透与氢气分压差的关系,在膜两侧分别通入氢气与氮气,并在两种不同氮气压力条件下进行测量(0.1与0.5 MPa).如果压差能够作为气体透过膜的驱动力,则膜在压差下的氢渗透率将大于平衡压力下的氢渗透率.但从结果可以看出,0.1与0.5 MPa的不同氮气压力条件下通过膜的氢渗透量相等,如图6所示.因此,在所考虑的压力范围内,压差作为气体以对流形式通过Nafion膜渗透的驱动力可忽略不计.这主要是由于当施加的压差低于Nafion膜水通道中的毛细管压力时,气体、液体通过Nafion膜水通道的传输不受压差驱动.这也表明了在该条件下,通过Nafion膜的氢渗透具有扩散性质,即以布朗运动溶解的气体形式通过膜渗透.根据式(4),渗透与氢气分压差的线性关系也说明了渗透率在0.5 MPa的压力范围内保持恒定,通过渗透率与扩散系数、溶解度的关系式ε=DS,印证了扩散系数D与溶解度S在0.5 MPa范围内不随压力变化,与文献[7]结果一致.

图6 通过Nafion117膜的氢气渗透通量与氢气分压的关系曲线[14]Fig.6 Measured hydrogen permeation flux density through a Nafion N117 membrane as a function of partial hydrogen pressure[14]
4.2 渗透与氢气分压差的非线性关系
Schalenbach等[14]开展的氢气渗透研究是在渗透池环境中进行的,为无电流状态,结果显示了渗透通量随氢气分压差的线性关系.但在电解制氢过程中,存在电流的影响,基于此,Trinke等[28]研究了电解制氢实际环境中氢气渗透通量( ΦH2)随氢气分压差(ΔP)的变化规律,结果如图7所示.可以看出,在四种不同温度条件下,氢气渗透通量与分压差的关系均表现出二次相关性.

图7 不同温度下电解池中氢气渗透通量与分压差的关系曲线(阳极压力为0.1 MPa)[28]Fig.7 Hydrogen permeation flux as a function of a pressure difference for a PEM electrolyzer cell at asymmetric pressure and at different temperatures (pa=0.1 MPa) [28]
基于渗透与分压差的线性关系[19,23],许多研究得出氢气渗透为纯扩散性质的结论,但该研究结果显示出无法用纯扩散描述的二次相关性.据此,作者在扩散模型中扩展了对流传输,以解释二次相关性,结果如图8所示.可以看出,纯扩散渗透(, 图8虚线)仅适用于低压差范围,当压差在0.5 MPa以下时,对流渗透(,图8点线)所占的比重很低,整体仍表现出扩散性质;而当压力差进一步升高时,由于对流渗透与压力差的二次相关性,对流在整体渗透中占的比重越来越大.

图8 60 °C条件下氢气渗透通量与压力差的关系曲线 [28]Fig.8 Hydrogen permeation flux at 60 ℃ as a function of pressure difference [28]
作者进一步分析了对流渗透形成的原因,对于电解制氢过程中的对流氢传输,需要高透水率促使水从阴极流向阳极.可能的机制包含两个方面:一是膜的透水性随离子交换当量(EW值)的降低而提升[29],与 Nafion117(EW 值为 1100 g·mol−1)相比,该研究所用膜的离子交换当量要低的多(EW 值为 910 g·mol−1),约 200 g·mol−1的差异可能会使透水性大幅提高.另一方面的原因在于操作条件的影响,如图9(a)所示,该研究是在电解制氢工作状态下进行的,电渗作用会促使水从阳极传输至阴极[30−32],导致膜中的水通道变宽,同时使得阴极侧的膜/水界面由疏水性转变为亲水性,降低了界面传输阻抗,从而有助于水在压力差作用下由阴极流向阳极,形成对流渗透.
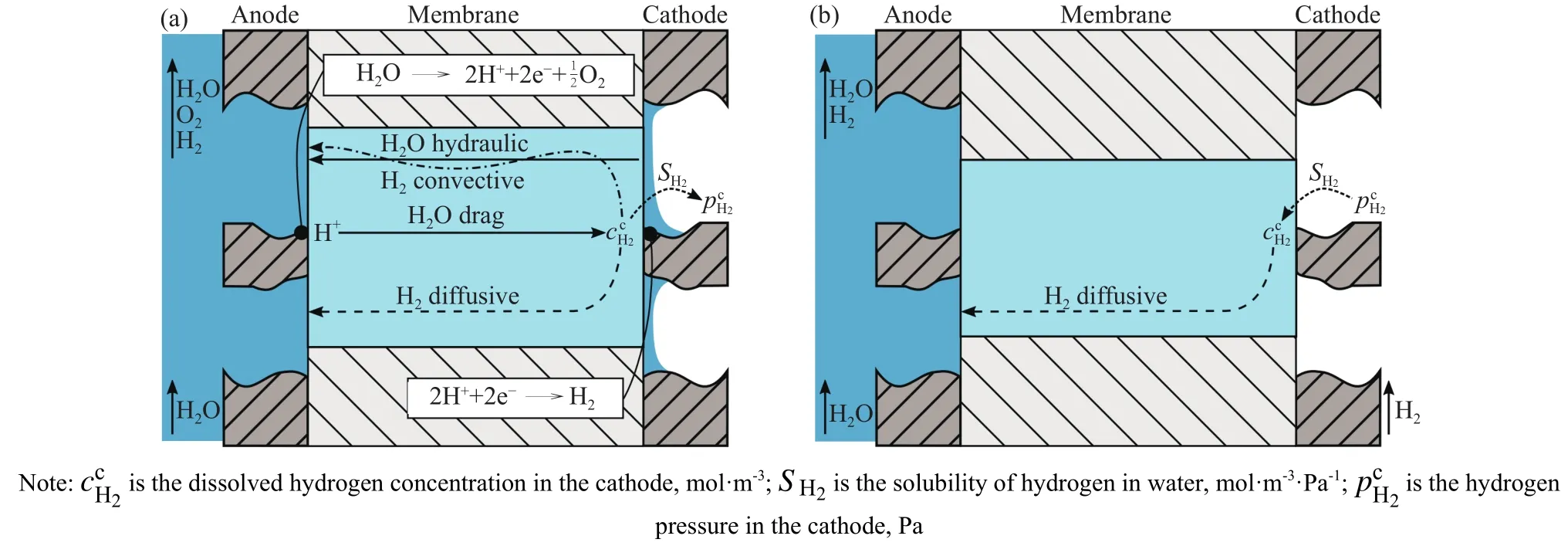
图9 测量条件示意图.(a) 水电解条件下测量; (b) 渗透池条件下测量(未施加电流)[28]Fig.9 Schematic measurement conditions for (a) measurement during electrolysis and (b) measurement in a permeation cell without applying current[28]
5 电流密度对渗透的影响
Trinke等[33]研究了电解制氢运行电流密度对氢气渗透的影响,该研究是在EF−40膜、0.05~1 A ·cm−2、30~80 °C、0.1~3.1 MPa阴极压力条件下进行的,图10示出了80 °C、阴极压力为0.1 MPa条件下的结果.从图10(a)可以看出,氧中氢体积分数(φH2)随运行电流密度的变化显示了双曲线的特征趋势,主要是由于析氧量随运行电流密度升高而增加.在低电流密度下,阳极的氢含量非常高,这对于控制制氢系统的安全十分关键.将氧中氢体积分数换算为氢渗透通量(ΦH2)与渗氢电流密度(IH2),结果如图10(b)所示,氢渗透通量与渗氢电流密度随运行电流密度(I)增大呈线性增加的趋势.此外,图10中同时列出了其他文献的研究结果[34−35],尽管所采用的膜与测试条件并不完全一致(文献 [34]的条件:Nafion 117 膜,T=80 °C, 阴极压强pc=0.7 MPa;文献 [35]的条件: Nafion 117 膜,T=85 °C, 阴极压强pc=0.1 MPa),但氧中氢体积分数与氢渗透通量随运行电流密度均表现出类似的变化规律.
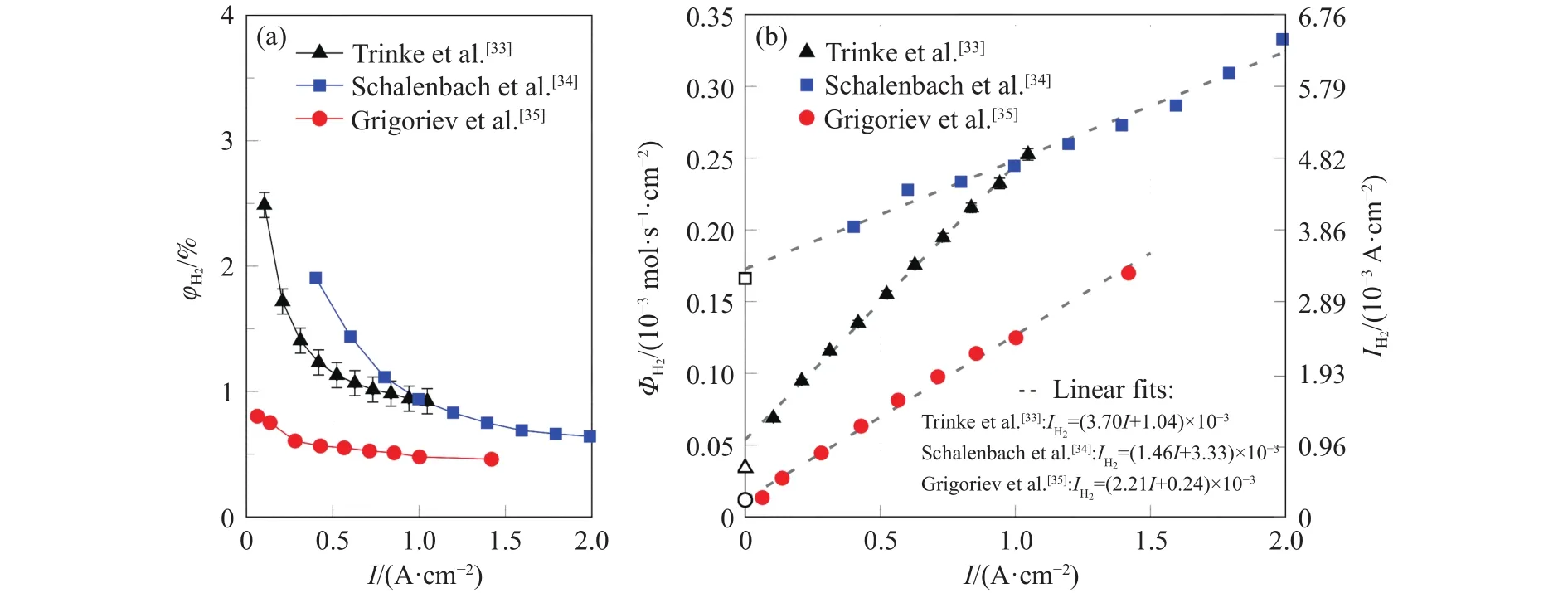
图10 不同文献中运行电流密度对氢气渗透影响的关系曲线对比.(a)氧中氢体积分数; (b) 氢气渗透通量[33-35]Fig.10 Comparison of effects of current density on hydrogen permeation: (a) hydrogen volume fraction; (b) hydrogen permeation rate[33-35]
研究结果显示随运行电流密度增大,氢渗透通量增加.但考虑到电解制氢工作条件,高电密下电渗作用引起的水通量可能导致溶解的氢由阳极传输回阴极,则电流密度增大可能导致氢渗透率降低.针对该问题,作者对比分析了电流密度升高对渗透可能产生的影响机制,包括催化层局部压力升高[36−39]、局部温度升高[40−41]、膜中水通道结构改变[27]和氢过饱和[42−48].其中,氢过饱和理论能够较合理地解释渗透通量随电流密度增大的规律,即阴极催化层的离聚物内溶解的氢达到过饱和状态.如图11所示,假设分子氢在阴极催化层的水中首先以溶解氢的形式产生,从溶解氢转变为气态氢,需要经过路径A,一旦达到氢溶解度的最大值,便会发生路径A的各步骤( Ⅰ: 从催化剂解吸;Ⅱ: 通过离聚物传输;Ⅲ: 界面传输至液态水;Ⅳ:通过水传输;Ⅴ: 气泡形成/增长;Ⅵ: 界面传输至气相),但这种传质过程是受限的.因此,离聚物中溶解的氢浓度高于理论值,且施加的电流密度越高,溶解的氢含量越高,导致溶解氢的过饱和度升高.过饱和的氢将通过路径B渗透至阳极,最终导致氢渗透增加.
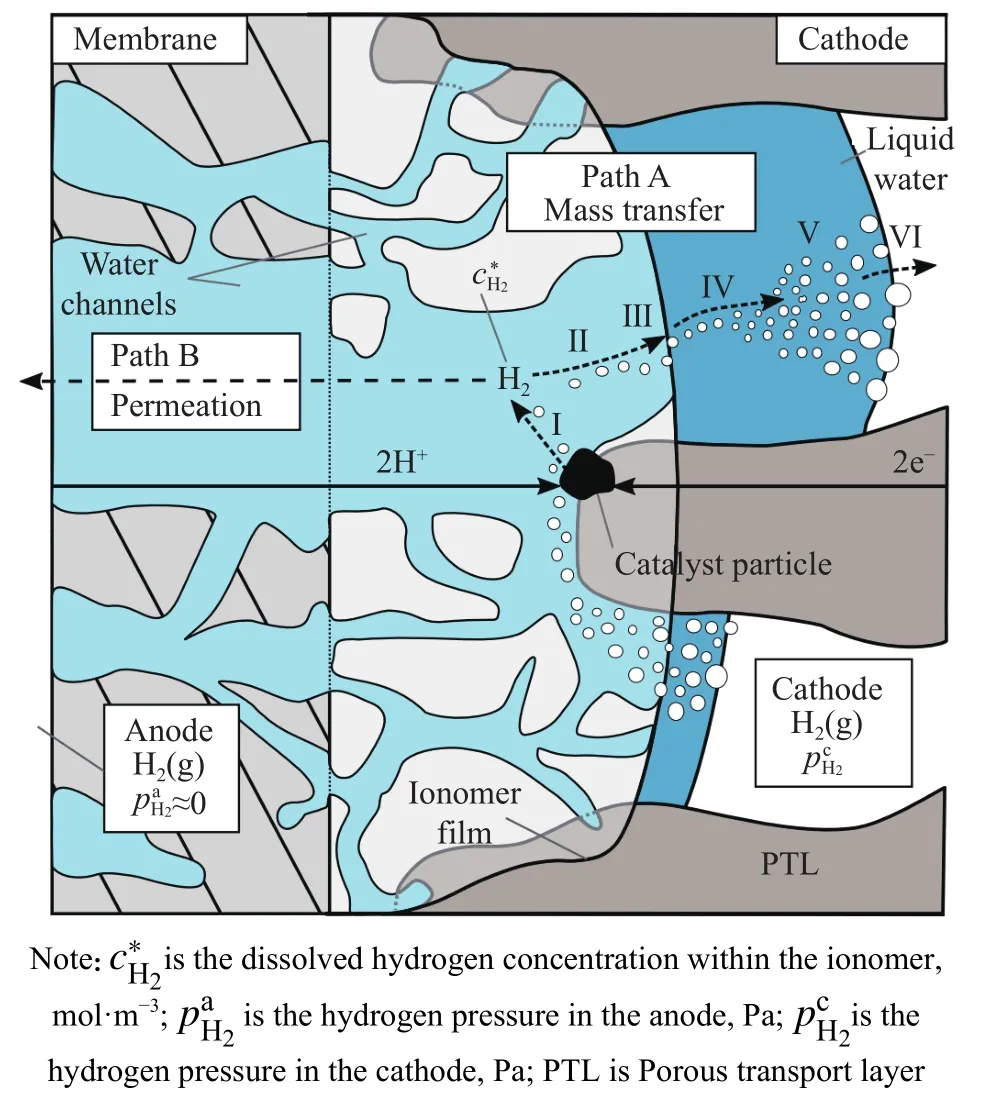
图11 阴极催化层离聚物中氢过饱和示意图[33]Fig.11 Sketch of the hydrogen supersaturation within the ionomer film of the cathode[33]
6 结论
目前,通过理论计算结合试验,对质子交换膜电解制氢的氢气渗透行为有了较深入的理解,不同运行参数对氢气渗透的影响规律已取得一定进展,但在相关影响机理方面仍未统一,需后续研究进一步探索与验证:
(1) 在质子交换膜电解制氢的常规运行压力范围(3.5 MPa)内,扩散系数与溶解度主要受温度影响,压力产生的影响很小,温度升高则渗透率增大;
(2) 氢气在水中的渗透率约为干膜的5~10倍,但不同相对湿度膜的氢气渗透研究表明,由于水与聚合物基质的相互作用,中间相与固相可能成为渗透的主体;
(3) 分压差对氢气渗透的影响表现出线性(渗透池环境)与非线性(电解制氢环境)两种关系,非线性可能源于膜透水性提升与水通道结构改变引起的对流渗透;
(4) 氢气渗透通量随电流密度升高而增大,氢过饱和是可能的影响机理,高电流密度下氢过饱和度升高,导致通过膜的渗透增加.
