枯叶蛱蝶中华亚种基因组Survey分析
2022-06-13洪芳徐堉峰范博
洪芳,徐堉峰,范博
(1.泉州师范学院 海洋与食品学院,福建 泉州 362000;2.台湾师范大学 生命科学系,台湾 台北 116)
枯叶蝶,学名枯叶蛱蝶(Kallimainachus),隶属于蛱蝶科Nymphalidae,是大型蝴蝶,以前后翅相叠其翅形及斑纹似枯叶而著称,具有较高的仿生学研究价值[1].枯叶蛱蝶[2]是枯叶蛱蝶属中分布最为广泛的物种,为该属的代表种.目前,枯叶蛱蝶已知有6个亚种 (不含海南亚种),包括指名亚种K.inachusinachus[2]、中华亚种K.inachuschinensisSwinhoe、台湾亚种K.inachusformosanaFruhstorfer、日本亚种K.inachuseucercaFruhstorfer、暹罗亚种K.inachussiamensisFruhstorfer和北印度亚种K.inachushuegeli(Kollar).枯叶蝶一年多代,成虫常栖息于阔叶林,出现在湿润繁茂雨林里的灌木丛中和河床两岸,在南方几乎全年可见[3].枯叶蛱蝶栖息于天然植被覆盖较好的山区,其幼虫寄主植物有荨麻科Urticaceae、蓼科Polygonaceae、蔷薇科Rosaceae和爵床科Acanthaceae[4];在我国仅记载有爵床科Acanthaceae芦莉花族Ruellieae、鳞花草族Lepidagathideae和爵床族Justicieae的部分植物种类[5].幼虫能以爵床科多种马兰属植物为寄主.成虫则以腐烂水果、阔叶树干虫蛀伤口流出的树液和动物粪便为食,是典型的食腐蝶类.垂直栖息海拔根据环境而定,一般可达到1 800 m的高度[6],尽管Mark Alexander Wynter-Blyth记录了它在海拔2 400 m森林茂密的山区和丘陵地区会遇到[4].在喜马拉雅山脉印度库曼地区,枯叶蛱蝶栖息在400~1 400 m的热带落叶森林和1 200 m以上的亚热带常绿森林[6].1998-2004年在中国重庆市进行的一项调查发现,枯叶蝶栖息于潮湿阔叶林中[7].枯叶蛱蝶广泛分布于东南亚的国家和地区,如印度、尼泊尔、不丹、孟加拉国、缅甸、中国、泰国、老挝、越南、巴基斯坦和日本[6].在我国分布于除海南外的秦岭以南地区.
当前,第二代测序技术 (next-generation sequencing technologies,NGS)和PacBio的单分子实时(single molecular real time,SMRT) 测序技术被广泛开发和应用,并迅速占领基因组测序领域的主流行业.Survey评估中的K-mer分析能得到更为全面和准确的信息,是因为K-mer分析是从数学角度进行分析的,克服了传统Feulgen分光光度法[8]和流式细胞法用来测定基因组大小[9-10]的劣势.由于内参选择不同,实验评估基因组大小与实际会有一些偏差的不足.第二代测序技术因其快速发展及推广,进而加速了全基因组的研究进程[11].通过对枯叶蝶的全基因组测序,可为今后仿生学领域的研究提供科学理论依据.
1 材料与方法
1.1 材料
本研究所用材料为枯叶蛱蝶中华亚种,2017年6月购于四川省乐山市枯叶蝶养殖基地.所用活体样本带回实验室,液氮速冻后置于超低温冰箱(-80 ℃)保存,备用.
1.2 方法
1.2.1 基因组DNA提取、检测及测序 首先,采用改良的SDS法提取枯叶蛱蝶样本的基因组DNA,用紫外分光光度计 (日本岛津,UV-1800)检测浓度,电泳的Marker为1 kb DNA Ladder(TaKaRa)和λ-HindⅢdi-gest(TaKaRa,日本);然后,随机打断枯叶蝶的DNA样品,构建长度为250 bp的小片段文库,再通过末端修复、加测序接头盒A尾、PCR扩增、纯化等处理后完成文库制备.构建好的文库委托北京诺禾致源科技股份有限公司完成.利用Illumina Hiseq 2500测序平台进行双末端测序.为了保证分析数据的准确性,首先对测序产生的原始序列(raw reads)进行过滤,去除接头污染等低质量的片段,其后将得到的高质量序列(clean reads)用于每个物种的基因组大小预测以及杂合度和GC含量分析.
1.2.2 基因组特征预估 在基因组组装前,通过测序得到的序列估计基因组特征.采用基于K-mer的分析方法和结果来估计基因组大小和杂合率等,这里取K=17[12]来进行分析.
(1)根据Lander_waterman算法,基因组大小(G)满足如下公式:
Cbase=Ck-mer×L/(l-k+1),G=nk-mer/Ck-mer=nbase/Cbase.
其中:nbase和nk-mer为序列碱基总数和K-mer数,Cbase和Ck-mer为覆盖碱基的期望深度和K-mer期望覆盖深度.
(2)K-mer深度频率分布服从泊松分布:
其中:a1/2为杂合K-mer种类数的百分比,为所有K-mer的种类数.
1.2.3 基因组初步组装 通过所有小片段库测序得到的reads截断成更小的序列片段之间的重叠关系构建de Brujin图.去掉无法继续连接的分支、低覆盖度的分支,并且利用reads信息化简单重复序列在de Brujin图的分叉通路,才能简化非常复杂的de Brujin图.采用随机选择策略,合并少量的杂合位点,得到一个简化后的de Brujin图.只有在每个分叉位点将序列截断,才能得到最初的contigs,是因为简化后的de Brujin图仍然会有很多分叉位点无法确定真正的连接关系.根据插入片段大小信息和reads之间的连接关系,将所有文库测序得到的reads比对回初步得到的contigs,将contigs组装成scaffolds.构建contig 和scaffold采用K=41,进行SOAP de novo组装[13]要根据高质量数据,在得到scaffold序列后,要先用SOAP将过滤后的reads 比对到该组装序列上,再直接拼接,从而获得原始基因组序列及碱基深度.以10 kb为窗口,在序列上无重复前进,从而做出GC_depth点图.计算每个窗口的平均深度与GC含量,利用GC 聚成块的分层来判断基因组的杂合率和重复的分布情况.
2 结果与分析
2.1 测序数据量
使用第二代高通量测序技术测序,共获取28.25 Gb原始数据.若预计样品基因组大小为545.05 M,则枯叶蝶样品的测序深度为52×.先建立一个DNA文库, 插入片段大小350 bp,严格过滤后得到clean data.产出初始数据28 249 103 100 bp,过滤后28 183 572 900 bp,GC含量33.94%,测序错误率0.02%.
2.2 K-mer分析
枯叶蛱蝶的原始数据用于17-mer分析使用,得出其频率分布(图1).K-mer的总数是21 588 M.以17-mer出现的次数作为横坐标,出现的频率作为纵坐标,可以看出,17-mer分布曲线成峰情况较好.K-mer 深度分布受基因组中杂合子和重复序列存在的影响.因为K-mer曲线呈现明显拖尾(图1),说明枯叶蝶基因组重复序列含量较高.从survey分析的结果可以看出,在depth=39附近是主峰值,由K-mer-number/depth)得到的基因组大小为553.55 Mb左右,修正后的基因组大小为545.05 Mb(图2).经过计算和分析得出,枯叶蝶的基因组杂合率为1.22%,重复序列比例为48.03.%.

图1 K-mer=17 Depth和K-mer个数频率分布 图2 K-mer=17 Depth和K-mer种类数频率分布Fig.1 Frequency distribution of K-mer =17 Fig.2 The frequency distribution of K-mer=17 Depth and K-mer number Depth and K-mer species number
2.3 基因组初步组装
用K-mer 41进行初步组装,统计得到Contig N50为555 bp,总长为653 208 907 bp,Scaffold N50为972 bp,总长为708 125 193 bp(表1).由各分析指标来看,测序枯叶蝶基因组为复杂基因组,后续可以采用相应策略进行基因组组装.
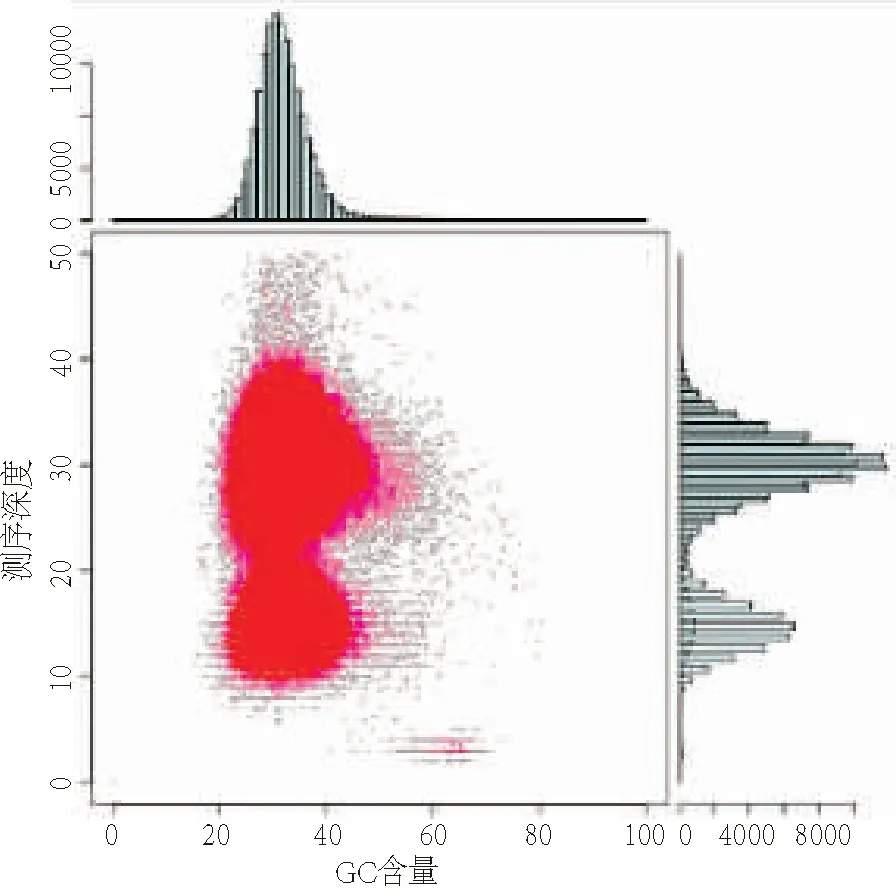
图3 contig GC含量和覆盖深度Fig.3 GC content and average sequencing depth of the Kallima inachus genome data 注: 红色的部分代表散点图中点的密度比较大的部分

表1 组装结果统计Tab.1 Statistics of assembly results in the Kallima inachus genome bp
2.4 GC含量及分布
在全基因组尺度上,基因组中的碱基组成,腺嘌呤(A)、鸟嘌呤(G)、胸腺嘧啶(T)、胞嘧啶(C)的相对含量,一般是用GC含量(GC-content)来表示[14-15].因GC含量的变化跟重复元件分布、基因密度、同义密码子的使用频率等密切相关,故其含量能体现生物体核酸序列组成的重要特征[16].
经过计算预估,枯叶蛱蝶基因组GC含量为35.24%.对组装的contigs进行GC含量统计的结果见图3.由图中可知,枯叶蛱蝶的GC深度分布被分成了2层,出现了一个低深度区域,基因组的杂合率(1.97%)高是可能的原因.因为杂合会导致两条同源染色体杂合的部位只组装出了1条,或2条都没有装出;同时,GC含量出现较低的一层,正是由于该部位以上的read乘数是整个基因组乘数的一半[17].根据图3中GC含量的深度信息,发现GC图发生轻微的分离现象;根据验证比对后推测,枯叶蛱蝶样品中含有外源污染.
2.5 蛱蝶科昆虫基因组比较
与本文枯叶蝶进行比对的有33种蛱蝶的基因组组装信息,这些蝴蝶的基因组组装信息来自于NCBI-genome (https://www.ncbi.nlm.nih.gov/genome).检索得到的33 条蛱蝶科昆虫相关的全基因组信息见表2.由表中可知,尚有喙蝶亚科Libytheinae、螯蛱蝶亚科Charaxinae、闪蝶亚科Morphinae、绢蛱蝶亚科Calinaginae、釉蛱蝶亚科Heliconiinae、线蛱蝶亚科Limenitidinae、丝蛱蝶亚科Cyrestinae、苾蛱蝶亚科Biblidinae和小紫蛱蝶亚科Apaturinae等8亚科未见全基因组信息.在已报道的完成全基因组测序的8 种蛱蝶中,帕眼蝶(P.aegeria)的基因组为501.7 Mb,偏瞳蔽眼蝶(B.anynana)为475.4 Mb[24],金斑蛱蝶(H.misippus) 为408.79 Mb[19],黑妹袖蝶(Heliconiushimera)为399.74 Mb,Dionevanilla为390.75Mb[21],庆网蛱蝶(M.cinxia)为389.908 Mb[18],黑脉金斑蝶(D.plexippus)为241.87 Mb[20],红带袖蝶(H.melpomene)为239.614 Mb[22].我们测定的枯叶蝶的基因组大小为545.05 Mb,除略小于Maniolajurtina(614.74 Mb)和翠袖锯眼蝶(Elymniashypermnestra)(566.93 Mb)外,明显大于上述8种蛱蝶及其余蛱蝶的基因组.

表2 枯叶蝶基因组组装数据及与33个蛱蝶科昆虫基因组比较Tab.2 Genome assembly statistics of K.inachus and comparisons to 33 available genomes of Nymphalidae
枯叶蛱蝶基因组的GC 含量为35.24%(图3).枯叶蝶的GC含量低于蛱蝶科眼蝶亚科的Maniolahyperantus(37.89%)、帕眼蝶(37%)、偏瞳蔽眼蝶(36.6%)及Taenariscatops(36.6%),高于其他29种蝴蝶的GC 含量(表2).已有研究表明GC 含量存在种的差异[25],在蛱蝶科GC含量在30%~38%(表2). 如果GC 含量过高(>65%)或过低(<25%),都可能导致Illumina 测序平台上的序列偏倚,并严重影响基因组装配[26].
3 讨论
在全基因组测序中,一般要求Contig N50>20 kb,Scaffold N50>300 kb,才能拼接出比较满意的基因组序列[17].二代测序技术中常用的组装软件包括 GS Assembler、ABySS、Velvet和SOAP 等.为了克服单个拼接软件分析结果精确度的局限性,我们使用了多种序列的拼接软件用于枯叶蛱蝶测序结果的拼接中,这样序列拼接的精确度才能得到显著的提高.这对于众多的非模式昆虫全基因组测序研究具有一定的借鉴意义.枯叶蛱蝶基因组的杂合率为1.22%,说明其基因组具有较高杂合率,其杂合率远远高于同科著名的迁徙性蝴蝶黑脉金斑蝶(0.55%).这有待于今后的进一步研究探讨.
本研究经过对枯叶蛱蝶基因组大小的首次测定及相应参数的初步评估,现得出如下结论:(1)目前枯叶蛱蝶基因组大小可以粗略估计为545 Mb;(2)通过对枯叶蛱蝶基因组28.25 G测序数据采用K-mer17进行survey分析,预估得到基因组大小为553.55 Mb,修正后为545.05 Mb,重复序列比例为48.03%.因为枯叶蛱蝶有较高的杂合度和重复,由各种分析指标来看,可以推测该测序枯叶蛱蝶基因组为复杂基因组范畴,但用全基因组鸟枪法策略进行组装还是存在一定的风险和难度.
