New insights in diagnosis and treatment of gastroenteropancreatic neuroendocrine neoplasms
2022-06-11FengYinZiHaoWuJinPingLai
Feng Yin, Zi-Hao Wu,Jin-Ping Lai
Abstract Gastroenteropancreatic neuroendocrine neoplasms (GEP-NENs) are rare epithelial neoplasms derived from pluripotent endocrine cells along the gastrointestinal tract and pancreas. GEP-NENs are classified into well-differentiated neuroendocrine tumors and poorly differentiated neuroendocrine carcinomas. Despite overlapping morphological features, GEP-NENs vary in molecular biology, epigenetic, clinical behavior, treatment response, and prognosis features and remain an unmet clinical challenge. In this review, we introduce recent updates on the histopathologic classification, including the tumor grading and staging system, molecular genetics, and systemic evaluation of the diagnosis and treatment of GEP-NENs at different anatomic sites, together with some insights into the diagnosis of challenging and unusual cases. We also discuss the application of novel therapeutic approaches for GEP-NENs, including peptide receptor radionuclide therapy, targeted therapy, and immunotherapy with immune checkpoint inhibitors. These findings will help improve patient care with precise diagnosis and individualized treatment of patients with GEP-NENs.
Key Words: Gastroenteropancreatic neuroendocrine neoplasms; Neuroendocrine tumours;Neuroendocrine carcinoma; World Health Organization classification; Diagnosis;Treatment
INTRODUCTION
Gastroenteropancreatic neuroendocrine neoplasms (GEP-NENs) are epithelial neoplasms with neuroendocrine differentiation that occur inside the gastrointestinal (GI) tract and the pancreas[1 ]. They are a group of tumors with significant heterogeneity and complex clinical behavior, ranging from slowly growing well-differentiated gastroenteropancreatic neuroendocrine tumors (GEP-NETs) to highly aggressive, poorly differentiated gastroenteropancreatic neuroendocrine carcinomas (GEP-NECs)[1 -3 ].The traditional term for a NET is carcinoid, which has been largely discouraged in the updated disease classification published by the World Health Organization (WHO)[1 ].
GEN-NENs are relatively rare (1 .0 %-1 .5 % of all GEP neoplasms, 6 .98 and 0 .4 new casesperyearper100000 individuals in the United States for GEP-NETs and GEP-NECs, respectively)[4 -7 ], although their incidence has significantly increased in the past 3 decades, largely due to the improved awareness and detection rate. The majority (> 95 %) of GEP-NENs are sporadic, although some (approximately 5 %)could be part of syndromic presentations, including multiple endocrine neoplasm type 1 (MEN1 ),neurofibromatosis type 1 (NF1 ), and von Hippel-Lindau syndrome (VHL)[1 -4 ].
Based on the embryological development of the GI tract, GEP-NENs can be divided into foregut (the esophagus to the proximal duodenum and pancreas), midgut (the distal duodenum to proximal two thirds of the transverse colon), and hindgut NENs (distal third of the transverse colon to the rectum)[5 ,6 ], with the midgut (especially the small intestine) being the most common site for GEP-NENs. For example, Figure 1 shows the case of a 71 -year-old Caucasian man who was initially identified to have multiple liver masses on computed tomography (CT) scans. CT-guided percutaneous liver needle core biopsy showed poorly differentiated neuroendocrine carcinoma and a small cell type, and the tumor cells were positive for neuroendocrine markers and GI tumor markers. A positron emission tomography(PET)/CT scan revealed the primary lesion in the ileum (Figure 1 ). In addition, compared with their foregut and hindgut counterparts, midgut NENs are more commonly associated with carcinoid syndrome[7 ]. In this review, we will discuss anatomic origin and pathologic feature-based classification systems for GEP-NETs as well as the diagnosis and current update of therapy. Based on our practice, we will also share some experience in the work-up of some unusual and challenging cases with diagnostic pitfalls.
HISTOPATHOLOGIC CLASSIFICATION
Tumor differentiation is closely associated with the clinical behavior of GEP-NENs and refers to how much the tumor tissue looks like the normal tissue that it was derived from. Based on tumor differentiation and histopathologic features, GEP-NENs can be classified into three major categories: Welldifferentiated NETs, poorly differentiated NECs, and mixed neuroendocrine-nonneuroendocrine neoplasms (MiNENs)[8 ]. Well-differentiated GEP-NETs commonly present with a uniform population of tumor cells with round nuclei and finely stippled “salt-and-pepper” chromatin[9 ]. Their common growth patterns include nests, trabeculae, acini, and ribbons. On the other hand, poorly differentiated GEP-NECs could be further classified into small-cell NECs and large-cell NECs based on their cytological features and commonly grow in sheets or nests with frequent tumor necrosis[10 ]. Small-cell NECs have features that include blue cells with scant cytoplasm, finely dispersed chromatin, nuclear molding, smudging, no distinct nucleoli, high mitotic rate, and patterns of rosettes and/or peripheral palisading (Figure 1 ). Large-cell NECs also have a neuroendocrine architecture and features of large cells with abundant cytoplasm, round and vesicular nuclei, and prominent nucleoli. Some NECs have a concurrent adenocarcinoma component and are categorized as MiNENs[11 ,12 ].

Figure 1 Ileal small cell neuroendocrine carcinoma with liver metastases in a 71 -year-old man. A: Computed tomography (CT) image (axial)showing ileal and multifocal liver lesions (arrow); B: Positron emission tomography-CT image showing ileal (arrow) and liver lesions (arrowheads) with hypermetabolic activity; C: Histopathologic features of small cell neuroendocrine carcinoma. Note the tumor cells with peripheral palisading, rosetting, scant cytoplasm, nuclear molding, finely granular chromatin, and lack of prominent nucleoli; D-F: Tumor cells with positive immunoreactivity for synaptophysin (D) and CDX2 (E) as well as a high Ki-67 proliferation index (70 %) (F). (C-F: 400 ×).
Tumor grade is another important factor closely correlated with the clinical behavior of GEP-NENs. It refers to how abnormal the tumor cells look under a microscope, and in the case of GEP-NENs, the tumor grade is usually determined by the proliferation rate of the tumor cells that could be reported by the mitotic rate (number of mitosesper2 mm2 ) and/or the Ki-67 proliferation index (average nuclear immunolabeling based on at least 500 tumor cells) (Table 1 ). The current guidelines use a 3 -tier tumor grading system: Low-grade (grade 1 , G1 ) tumors with a mitotic rate up to 2 per 2 mm2 or a Ki-67 proliferation index up to 3 %, intermediate-grade (grade 2 , G2 ) tumors with a mitotic rate from 2 to 20 per 2 mm2or a Ki-67 proliferation index from 3 % to 20 %, and high-grade (grade 3 , G3 ) tumors with a mitotic rate greater than 20 per 2 mm2 or a Ki-67 proliferation index greater than 20 %[13 ]. A suggestion has been made to use a Ki-67 proliferation index of 5 % as the cutoff level, instead of 3 % according to current guidelines, for better risk stratification in patients with G1 to G2 tumors[14 ], although additional largescale studies are needed to validate this proposed cutoff value. Due to the heterogeneity among tumor tissues, a routine practice is to perform measurements in the most mitotically active tumor area. In cases with discrepancies between the mitotic rate and Ki-67 proliferation index, the tumor will be placed into the highest-grade category. A higher Ki-67 proliferation index is associated with a poorer prognosis[15 ].In fact, the Ki-67 proliferation index appears to be a better prognostic marker than the mitotic rate for metastatic pancreatic and midgut NENs[16 ]. Of note, all NECs were high-grade carcinomas with a poorly differentiated morphology and a high Ki-67 proliferation index (> 20 %, more than 50 % in the majority of the cases) (Figure 1 ) and high mitotic count (> 20 per 2 mm2 ). Historically, all G3 GEP-NENs were conceptually equal to NECs before 2017 . However, recent studies have clearly demonstrated that G3 GEP-NETs and GEP-NECs are genetically different entities[17 ]. In general, G3 GEP-NETs are morphologically well differentiated and clinically less aggressive than GEP-NECs and have a poorer response to platinum-based chemotherapy[18 ].
All GEP-NENs are characterized by the expression of neuroendocrine markers, with or without secretion of biologically active substances. Immunohistochemical staining is often necessary, not only to confirm the diagnosis and to assign the tumor grade category but also to investigate the tumor origin in cases of metastasis. GEN-NENs are derived from the neuroendocrine epithelium and therefore normally express cytokeratin (CK), with CK8 and CK18 being the most common[19 ]. The expression of CK could separate GEP-NENs from their great mimics pheochromocytoma/paraganglioma. General neuroendocrine markers are also frequently used in routine practice to establish the diagnosis. Well-differentiated NETs usually express somatostatin receptors. In fact, the expression of somatostatin receptor subtype 2 A (SSTR2 A) is the basis of functional imaging (such as gallium Ga 68 dotatate) and somatostatin analog (SSA) therapy, including octreotide acetate and peptide receptor radionuclide therapy (PRRT) (such as lutetium Lu 177 dotatate)[20 ]. Some commonly used neuroendocrine immunohistochemical markers include chromograin A (CgA), synaptophysin (SYN), and CD56 . Recent studies have demonstrated INSM1 (insulinoma-associated protein 1 ) as a novel and more specific marker ofneuroendocrine differentiation. In the case of poorly differentiated NECs, INSM1 appears much more sensitive (95 %) than CgA (83 %) and SYN (82 %)[21 ]. Immunohistochemical staining could also be helpful to identify unknown primary tumors in cases of metastasis. Up to 20 % of NETs originally present as liver or bone metastasis from unknown primary tumors, and identification of the primary tumor has significant therapeutic and prognostic implications. The jejunum, ileum, and pancreas appear to be the most common primary sites for patients with NET liver metastases of occult origin[22 ,23 ].CDX2 immunoreactivity is present in the majority of jejuno-ileal NETs, and up to 24 % of metastases are primarily pancreatic NETs[24 ]. The novel marker SATB2 (special AT-rich sequence-binding protein 2 ) is frequently and strongly expressed in NETs of the lower GI tract and has shown value in assigning NEC sites of origin[25 ]. For metastatic GEP-NENs, additional immunohistochemical panels include PDX-1 (pancreatic and duodenal homeobox 1 ), PAX6 (paired Box 6 ), PAX8 (paired Box 8 ), ISL1 (islet 1 ),NESP55 (neuroendocrine secretory protein 55 ), PR (progesterone receptor), and PrAP (prostate acid phosphatase)[26 ,27 ]. We performed PAX6 and PAX8 immunohistochemical staining on 178 NETs,including 110 primary NETs (26 pancreatic, 10 gastric, 12 duodenal, 22 jejuno-ileal, 10 rectal, and 30 pulmonary) and 68 NETs metastatic to the liver (24 pancreatic, 1 duodenal, 37 jejuno-ileal, 1 rectal, and 5 pulmonary). Among primary GEP-NETs, PAX6 and PAX8 were positive in 65 % (17 /26 ) and 73 %(19 /26 ) of pancreatic, 0 % (0 /10 ) and 10 % (1 /10 ) of gastric, 92 % (11 /12 ) and 92 % (11 /12 ) of duodenal,0 % (0 /22 ) and 0 % (0 /22 ) of jejuno-ileal, and 90 % (9 /10 ) and 80 % (8 /10 ) of rectal NETs, respectively.PAX6 and PAX8 positivity was seen in 46 % (11 /24 ) and 50 % (12 /24 ) of metastatic pancreatic NETs to the liver, respectively. None of the nonpancreatic NETs metastatic to the liver were immunoreactive for either PAX6 or PAX8 [27 ].
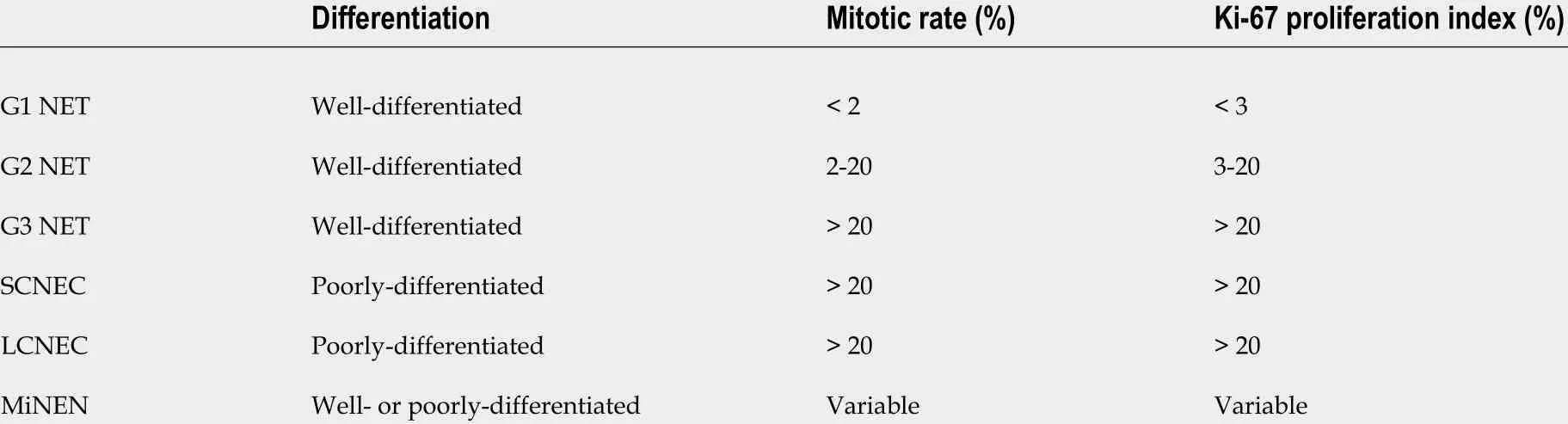
Table 1 World Health Organization classification for gastroenteropancreatic neuroendocrine neoplasms1
MOLECULAR GENETICS
GEP-NENs consist of a biologically distinct group of tumors with great genetic heterogeneity. Largely driven by high-throughput technologies and next-generation sequencing, significant progress has been made in recent years in understanding the key molecular drivers of tumorigenesis and progression,especially pancreatic and small bowel NENs[28 -30 ]. Approximately 10 %-20 % of pancreatic NETs (PNETs) are associated with hereditary genetic syndromes, including MEN-1 , NF1 , VHL, and tuberous sclerosis[31 ]. In cases with sporadic P-NETs, three types of major molecular alterations have been detected, including somatic mutations inMEN1(44 %),DAXX(death-domain associated protein)/ATRX(alpha thalassaemia/mental retardation syndrome X-linked mutations) (43 %), and mammalian target of rapamycin (mTOR) pathway genes such asPTEN,TSC2, and PIK3 CA (14 %)[32 ]. Identification of mTOR pathway gene alterations has clinical relevance due to the available targeted therapy. Germline mutations in DNA repair genes, includingMUTYH,CHEK2, and BRCA2,have been reported in sporadic P-NETs[33 ]. Chromosome 18 deletion is detected in 60 %-90 % of small intestinal NETs (SINETs), although its significance is still unclear at this time[34 ]. Chromosome 14 gain is also frequently detected in advanced and metastatic disease[35 ]. Approximately 8 % of SI-NETs have somatic mutations inCDKN1B[36 ,37 ]. A recent study demonstrated that, as epigenetically dysregulated tumors, SI-NETs could be divided into three subgroups: (1 ) Chromosome 18 deletion with CDKN1Bmutations and CpG island methylator phenotype (CIMP) negativity, the largest subgroup (55 %) with the most favorable prognosis; (2 ) The absence of arm-level copy-number variation (CNV) with a high level of CIMP positivity, the subgroup (19 %) with intermediate prognosis; and (3 ) The presence of multiple CNVs, the subgroup (26 %) with a young age at onset and the worst prognosis[38 ]. In addition to P-NETs and SINETs, more studies are needed to understand the molecular genetics of GEP-NETs in other anatomic sites.
From a molecular genetics point of view, pancreatic NECs (P-NECs) are entirely biologically different entities from P-NETs. The most common molecular alterations in P-NECs are somatic mutations inTP53 ,RB1, CDKN2 A, and KRAS[39 ]. Notably, mutations in the TP53 and RB1 genes appear to be recurrent molecular events in GEP-NECs from different anatomic sites, including the stomach and colorectum[40 ].KRASmutations have also been detected in gastric and colorectal NECs, althoughBRAFmutations have only been reported in colorectal NECs[30 ]. Interestingly, somatic mutations inDAXX,ATRX,andMEN1are almost exclusively detected in well-differentiated NETs but not poorly differentiated NECs[41 ].
The pathological diagnosis of grade-3 well-differentiated NETs and poorly differentiated NECs could be challenging in some cases because of the high mitotic rate (> 20 per 2 mm2 ) and high Ki-67 proliferation index (> 20 %), particularly for the limited sample made from endoscopic ultrasound-guided fine-needle aspiration (EUS-FNA). The distinct molecular profile could help to separate these two entities. In practice, immunohistochemistry using antibodies against DAXX, ATXR, p53 , and RB1 could be performed to surrogate for genetic status[42 ]. Poorly differentiated NECs frequently have absent RB1 and aberrant p53 protein expression, together with normal expression of DAXX and ATRX. On the other hand, normal RB1 and p53 protein expression is normally found in well-differentiated NETs.
CLINICAL PRESENTATION, DIAGNOSIS, AND TREATMENT
Gastric NENs
Gastric NENs (G-NENs) originate from different neuroendocrine cell types in the gastric mucosa,including enterochromaffin (EC) cells (serotonin producing), enterochromaffin-like (ECL) cells(histamine producing), D-cells (somatostatin producing), and G-cells (gastrin producing)[43 ]. The diagnosis of G-NENs is usually performed incidentally during upper GI endoscopy due to the lack of specific symptoms, although rare cases could be seen in systemic syndromes, especially Zollinger-Ellison syndrome. Gastric NETs (G-NETs) are commonly subclassified into three distinct types that are mostly derived from ECL cells (Table 2 ). NETs derived from D cells, G cells, and EC cells are extremely rare.
Type 1 G-NETs are the most common NETs (80 %-90 %) in the stomach and associated with advanced autoimmune metaplastic atrophic gastritis. It is more commonly seen in females that frequently have additional autoimmune disorders, such as type 1 diabetes mellites and Hashimoto’s thyroiditis. The presence of autoimmune antibodies, including anti-parietal cell antibodies and anti-intrinsic factor antibodies, leads to the destruction of parietal cells and achlorhydria[44 ]. Laboratory testing shows elevated serum gastrin, decreased vitamin B12 , and high gastric pH (> 7 ). Gastrin induces ECL cell hyperplasia (< 0 .5 mm) and ultimately G-NETs when the lesions measure 0 .5 mm or larger.
Type 1 G-NETs are usually diagnosed under upper GI endoscopy with biopsy. It usually presents with multiple small (< 1 cm) reddish polyps or nodules of the gastric body (Figure 2 A) and fundus.Histologically, type 1 G-NETs show tumor cells with abundant eosinophilic cytoplasm, monotonous round nuclei, and characteristic chromatin arranged in trabecular or nested patterns (Figure 2 B and C).Necrosis is not a feature for this type of tumor. The background gastric mucosa shows atrophic gastritis with frequent intestinal metaplasia (Figure 2 B and C inset). In addition, the spindle cell morphology of type 1 G-NETs has been reported by us with histological features mimicking spindle cell gastrointestinal stromal tumors (GISTs) (Figure 2 D)[45 ] and therefore represents a potential diagnostic pitfall.
Most type 1 G-NETs are small G1 tumors and are limited to the mucosa and rarely the submucosa.Imaging study is usually unnecessary. However, EUS is likely warranted if the tumor is greater than 1 -2 cm due to a higher risk for lymph node metastasis (2 %-9 %), muscularis propria invasion, and angioinvasion[46 ]. The management of type 1 G-NET is generally conservative with endoscopic surveillance due to its favorable prognosis. Endoscopic resection could be performed on cases with larger lesions (>5 mm) (Figure 3 ). Gastrectomy is only reserved for rare high-risk cases.
Type 2 G-NETs are rare and account for 5 %-6 % of G-NETs. They occur in Zollinger-Ellison syndrome in the setting of MEN-1 syndrome, and patients are younger. Type 2 G-NETs are usually caused by duodenal gastrinoma[47 ]. The common clinical presentation includes abdominal pain and watery diarrhea. Laboratory testing shows elevated serum gastrin and low gastric pH (< 2 ). Additional genetic testing is recommended to confirm MEN-1 syndrome in suspicious cases. Endoscopically, type 2 GNENs present with multiple gastric polyps or nodules. Multiple gastric peptic ulcers are common findings. These polypoid lesions are usually larger but typically less than 2 cm. The microscopic features of type 2 G-NENs are similar to those of type 1 G-NENs, presenting as low-grade tumors (G1 tumors being the most common) in a background of ECL hyperplasia. However, background gastric mucosa in type 2 G-NETS demonstrates parietal cell hypertrophy/hyperplasia. Type 2 G-NETs are mostly limited to the mucosa and submucosa. It has a higher risk of lymph node metastases (up to 30 %) and therefore a slightly worse prognosis than type 1 G-NETs.
The clinical management of type 2 G-NETs also includes endoscopic surveillance, endoscopic resection, and rarely surgery. It is of clinical importance to locate and resect the primary gastrinoma.SSAs have been proposed in the treatment of type 2 G-NETs, although large-scale cohort studies are necessary for their validation.
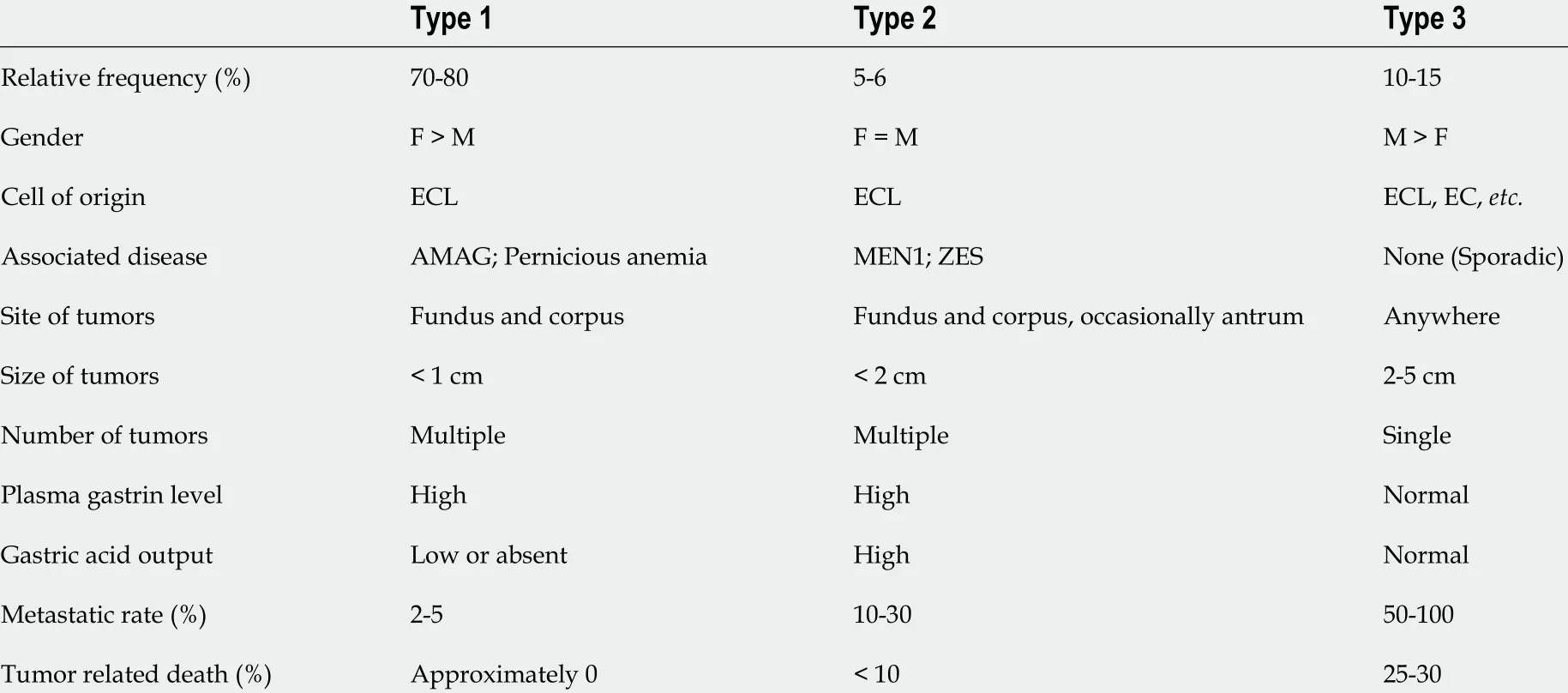
Table 2 Clinicopathologic characteristics of gastric neuroendocrine tumor
Type 3 G-NETs are sporadic tumors and account for 10 %-15 % of all G-NETs. Laboratory testing showed normal serum gastrin and gastric pH. The clinical presentations are nonspecific, including abdominal pain, melena, and weight loss. Carcinoid syndrome could be seen in patients with liver metastases. Endoscopically, the tumor is usually a single large lesion (> 2 cm) arising in the normal background mucosa. These tumors commonly have aggressive clinical behavior, characterized by higher tumor grade, higher tumor stage, and frequent lymph node and distant metastases. The diagnosis involves upper GI endoscopy and biopsy. EUS is required to measure the depth of invasion and to evaluate lymph node status. Additional imaging studies, including computed tomography (CT),magnetic resonance imaging (MRI), and somatostatin receptor scintigraphy (SRS), are recommended for perioperative tumor staging.
Compared with the favorable prognosis in types 1 and 2 G-NETs, type 3 G-NETs frequently present as high-grade and deeply invasive tumors. Lymph node metastases are found in up to 71 % of type 3 GNETs measuring 2 cm or larger[48 ]. Although endoscopic resection could be applied for small and superficial lesions, radical surgical resection (total or subtotal gastrectomy) with lymphadenectomy is often the treatment of choice for type 3 G-NETs.
Gastric NECs (G-NECs) are poorly differentiated carcinomas with high mitotic counts and frequent necrosis. They have been further subclassified into small cell NECs (SCNECs) and large cell NECs(LCNECs)[49 ]. Histologically, SCNECs are similar to their counterparts in the lung, featuring small neoplastic cells with scant cytoplasm and hyperchromatic nuclei. Prominent nucleolus is uncommon. In contrast, LCNECs have large neoplastic cells with abundant eosinophilic cytoplasm, vesicular nuclei,and prominent nucleoli. G-NECs are high-grade neoplasms by definition, with a high Ki-67 proliferation index (> 20 %, often more than 60 %-70 %) and a high mitotic rate (> 20 per 2 mm2 ). For G-NECs,surgical resection is often required, and postoperative chemotherapy is advised in cases with metastatic disease[50 ].
Small intestinal NENs
The incidence of small intestinal NENs (SI-NENs) has been increasing steadily over the past 3 decades to 1 .05 new cases per 100000 individuals per year[51 ,52 ], and they are the most common GEP-NENs(29 .5 %) in the United States, followed by the rectum (29 .2 %) and pancreas (13 .5 %)[52 ]. The clinical presentations include nonspecific abdominal pain, mass effects (small bowel obstruction), and symptoms related to excess hormone secretion. Ampullary NENs could cause jaundice and acute or chronic pancreatitis. Duodenal gastrinoma is a cause of Zollinger-Ellison syndrome. Despite relatively slow growth, the small intestine is the most common primary NET site for metastatic disease along the GI tract[53 ].
CT scans are the most common imaging modalities for the diagnosis of SI-NENs. Other imaging studies include ultrasound, MRI, and SRS. Endoscopy with biopsy is the gold standard for the diagnosis of SI-NENs. Endoscopic examination of SI-NENs includes capsule endoscopy, colonoscopy, and doubleballoon enteroscopy. Duodenal and periampullary NETs are usually single small (< 2 cm) polypoid or nodular lesions limited to the mucosa and submucosa. NETs at the jejunum and ileum are usually large(> 2 cm) and multifocal, with frequent deep invasion and lymph node metastases[54 ].
SI-NENs are morphologically similar to NENs at other sites. A few relatively specific histological features include psammoma bodies in somatostatin-producing D-cell NETs and nested growth patterns with peripheral palisading in serotonin-producing EC-cell NETs. Gangliocytic paraganglioma is a rare NET that is typically encountered in the second part of the duodenum and is characterized by the presence of three distinct components: A neuroendocrine epithelioid component, a Schwannian spindle cell component, and a ganglion cell-like component[55 ].
For localized SI-NETs, the standard of care is complete surgical resection of the primary tumor,regional lymph nodes, and mesenteric fibrotic tissue. A consensus has not been reached with routine administration of octreotide preoperatively or intraoperatively[56 ]. For metastatic disease, treatment options are surgical resection, liver-directed therapy (in cases predominantly with liver metastasis), and systemic therapy including SSA, PRRT, everolimus (mTOR inhibitor), and cytotoxic chemotherapy.
Appendiceal NENs
The appendix is a frequent primary site for GEP-NENs, with an incidence rate of approximately 0 .15 -0 .6 new casesper100000 individuals per year in the United States[52 ,57 ,58 ]. They frequently occur in children and young adults with a slight female predominance[59 ]. Appendiceal NENs (A-NENs) have the most favorable prognosis among all subgroups of GEP-NENs[52 ].
The common clinical presentations of A-NENs are similar to those of acute appendicitis. Carcinoid syndrome is extremely rare in A-NENs and is mostly associated with metastatic disease. Histopathologic evaluation is crucial to establish the diagnosis of A-NENs. The application of imaging studies,including CT and MRI, has limited value for the detection of small primary A-NENs. However,colonoscopy is recommended given that up to 18 % of patients with A-NENs have concurrent GI neoplasms[60 ].
The majority (80 %) of A-NENs are small and only incidentally found in appendectomy specimens[52 ]. Most A-NENs (60 %-75 %) are located at the appendiceal tip; therefore, the appendiceal tip should be examined carefully on all appendectomy specimens. The histological features of A-NENs are similar to those of NENs of other primary sites, with the exception of tubular NETs. Tubular NETs are rare benign neoplasms with a predominant tubular growth pattern, so it is important not to misdiagnose them as adenocarcinomas[61 ]. Of note, goblet cell adenocarcinoma (formerly goblet cell carcinoid) is no longer considered an A-NEN[62 ]. Currently, we believe that this is an unusual type of adenocarcinoma with neuroendocrine differentiation.
The management of A-NENs depends on the stage of the disease determined by the tumor size,location, and tumor extension. Simple appendectomy is considered adequate for tumors less than 10 mm. Right hemicolectomy is indicated for tumors larger than 20 mm. The implication of right hemicolectomy in A-NETs with a size of 10 -20 mm is still controversial, likely depending on the presence of high-risk features (positive margin after appendectomy, base location, Ki-67 index of 3 % or higher, > 3 mm mesoappendiceal invasion, angioinvasion, and perineural invasion)[63 ,64 ]. For patients with more advanced disease (stages III and IV), the treatment usually includes curative surgery and systemic therapy.
Colorectal NENs
The incidence rates of colonic and rectal NENs are 0 .2 and 1 .2 new cases per 100000 individualsperyear in the United States, respectively[52 ]. The mean age for colonic NENs is 65 years, which is significantly older than that for rectal NENs (56 years) due to late detection. The presentation of colorectal NENs is similar to that of colorectal adenocarcinoma with nonspecific mass-related effects, abdominal pain, and bleeding. Classic carcinoid symptoms could be seen in some cases, often with liver metastases.
The majority (70 %) of colonic NETs (C-NETs) are located on the right side of the colon, especially the cecum[65 ]. C-NETs are usually larger, with an average size of 4 .9 cm[66 ]. Approximately 30 %-40 % of CNETs have local or distant metastasis at the time of presentation. Colonoscopy with biopsy is commonly performed to establish the diagnosis. C-NETs are usually derived from EC cells or Kulchitsky cells within the crypts of Lieburkuhn. Therefore, C-NETs typically show EC cell features, including insular growth patterns and CDX2 immunoreactivity. Necrosis is usually absent. The preferred treatment is colectomy with lymphadenectomy.
Rectal NETs (R-NETs) are relatively smaller (< 1 cm), smooth, round polypoid lesions and generally have a better prognosis than their counterparts in the colon. R-NETs are subgrouped into the L-cell(glucagon-like peptide and pancreatic polypeptide producing) type and non-L-cell type according to their origin[67 ], with the L-cell type being the dominant type. L-cell R-NETs typically present with trabecular or tubular growth patterns. Of note, non-L-cell-type rectal NETs usually present as larger masses and have an increased risk of lymphovascular invasion and worse prognosis[68 ].
For the purpose of tumor staging, it is recommended to use EUS and MRI of the pelvis to determine the depth of invasion and lymph node status and to use SRS-based scans to determine distant metastases. Endoscopic mucosal resection and endoscopic submucosal dissection are indicated for small(1 cm or smaller) and superficial R-NETs if there is no evidence of muscularis propria invasion or lymph node metastases[69 ]. For R-NETs larger than 2 cm in size, low anterior resection or abdominal resection is recommended.
Pancreatic NENs
As rare neoplasms, the incidence rate for pancreatic NENs (P-NENs) is 1 .0 new cases per 100000 individualsperyear in the United States[70 ] and accounts for 2 %-4 % of all pancreatic neoplasms[71 ]. All P-NETs are considered to have malignant potential. These tumors are derived from pancreatic islet cells and could be subclassified into functioning and nonfunctioning subgroups. Functioning P-NETs,including insulinoma, gastrinoma, VIPoma, and glucogonoma, cause clinical hormone hypersecretion syndromes. The clinical presentations of functioning P-NETs are mostly related to hormone effects, such as hyperglycemia in insulinoma and large-volume secretory diarrhea in VIPoma. In contrast, nonfunctioning P-NETs are usually incidental findings on imaging studies for other causes or mass effects at late stages. With the increased use of imaging studies, nonfunctioning P-NENs have become more common,accounting for more than 60 % of all P-NENs[72 ].
Insulinoma and gastrinoma are the two most common functioning P-NETs. In suspected cases of insulinoma, 72 -h fasting tests for blood glucose, insulin, C-peptide, and proinsulin levels should be performed together with drug tests for sulfonylurea. In suspected cases of gastrinoma, laboratory testing includes fasting gastrin level and gastric pH. Laboratory testing for serum glucagon and VIP levels would be helpful for the diagnosis of glucagonoma and VIPoma. The circulating CgA level is also a sensitive and specific diagnostic marker for P-NETs, with the exception of insulinoma[73 ], and it has no added value for the diagnosis of nonfunctioning P-NETs.
The most common imaging studies for P-NENs include EUS, CT, MRI, and 68 Ga-dotatate PET.Radiolabeled glucagon-like peptide-1 receptor (GLP-1 R) scintigraphy is another sensitive tool to detect small insulinomas[74 ]. Based on our experience, P-NETs could appear as thin-walled cystic lesions with no communication with the pancreatic duct (Figure 4 ) that clinically and radiologically may mimic mucinous cystic neoplasms. Clinicians and pathologists should be aware of this unusual presentation to avoid misdiagnosis.
Microscopically, P-NETs are well-differentiated neoplasms that do not differ from NETs from other primary sites (Figure 5 ). One or more neuroendocrine markers (CgA, SYN, CD56 , and neuron-specific enolase) and one epithelial marker (cytokeratin AE1 /AE3 and CAM5 .2 ) are indicated for the diagnosis of P-NENs. Ki-67 immunoreactivity is warranted to assign tumor grade. Insulin immunoreactivity is necessary in the diagnosis of insulinoma in cases of multifocal tumors or insulinomatosis. In the cases of metastatic disease, some markers (Pax 6 , Pax8 , ISL-1 , PDX-1 , and CDX2 ) could be helpful to determine pancreatic origin[75 ].
Surgical resection is the preferred and only curative therapy for P-NENs. Conservative management was suggested for small (< 2 cm) low-grade nonfunctioning P-NETs due to their excellent prognosis[76 ,77 ]. However, even for small-sized nonfunctioning P-NETs, surgical resection is indicated in cases with high-risk features (55 years or older, grade 3 tumor, and distant metastases)[78 ]. Local resection or enucleation could be applied in localized and easily accessible disease to maximally reserve pancreatic tissue[79 ], especially if the tumor is located more than 2 -3 mm from the pancreatic duct. Depending on the tumor location, surgical resection procedures for P-NENs include partial pancreaticoduodenectomy and distal pancreatectomy. Regional lymphadenectomy is recommended for surgical resection of PNENs.
P-NENs commonly present with liver metastases. Surgical resection should be considered in metastatic disease for both nonfunctioning and functioning P-NENs. The presence of liver metastases is not a counterindication to surgical management for P-NEN patients[80 ]. Efforts should be made if surgical removal is feasible for both primary pancreatic tumors and metastatic liver lesions. However, it is still under debate whether to resect the primary tumor in cases of unresectable metastatic liver lesions. Surgical resection of metastatic liver lesions should be avoided in cases with unresectable primary P-NENs[81 ].

Figure 4 Pancreatic neuroendocrine tumor. A: Computed tomography (CT) image showing a 3 .5 cm distal pancreatic mass (arrow); B: Positron emission tomography-CT image showing a pancreatic mass with hypermetabolic activity (SUVmax = 4 .3 ) (arrow); C: Endoscopic ultrasound-guided fine-needle aspiration showing clusters of neuroendocrine tumor cells with round nuclei and fine stippled “salt-and-pepper” chromatin (H&E stain, 400 ×); D: Distal pancreatectomy showing the gross cut surface of a firm fibrotic pancreatic neuroendocrine tumor (T) with focal hemorrhage.
NOVEL THERAPEUTIC APPROACHES
The treatment options for advanced and metastatic GEP-NENs have significantly expanded during the past two decades[82 ,83 ]. Some important clinical studies, including the PROMID[84 ] and CLARINET[85 ] trials, have demonstrated a significant efficacy of SSA in the control of tumor growth in patients with metastatic GEP-NETs. A recent CLARINET FORTE phase 2 clinical trial further supports the clinical benefit of the SSA lanreotide autogel (LAN), which led to significantly improved progressionfree survival (PFS) and disease control rate in patients with GEN-NETs, especially in cases with a Ki67 index ≤ 10 %[86 ]. In addition to SSA[87 ], novel therapeutic approaches, including PRRT, targeted therapy, and immunotherapy, have demonstrated promising clinical benefits[88 -90 ].
PRRT is a type of systemic radiotherapy specifically targeting tumor cells expressing SSTR[91 ]. In the phase 3 NETTER-1 trial, for patients with metastatic well-differentiated midgut NETs, treatment with177 Lu-dotatate led to a significantly improved PFS (median PFS not reached vs 8 .4 mo in the control group with high-dose octreotide alone) and an improved radiographic response rate (18 % vs 3 % in the control group)[92 ]. The most common adverse effects for 177 Lu-dotatate are nausea and vomiting. Based on this trial, PRRT with177 Lu-dotatate has been approved for patients with advanced GEP-NETs and SSTR expression on imaging.
Due to the hypervascularity in GEP-NETs, multiple clinical trials have investigated the therapeutic effects of targeted therapy against vascular endothelial growth factor (VEGF) receptors. In a phase 3 trial, patients with low- to intermediate-grade P-NETs received placebovssunitinib, a tyrosine kinase inhibitor targeting multiple receptors, including VEGF receptors-1 , 2 , and 3 . Sunitinib led to a significantly longer median PFS [11 .4 mo vs 5 .5 mo in the control group; hazard ratio (HR) for progression or death, 0 .42 ; P < 0 .001 ][93 ]. Sunitinib has been approved for patients with advanced PNETs.
mTOR is a multifunctional serine/threonine kinase related to NET growth. mTOR pathway genes,includingPTEN,TSC2, and PIK3CA, are also frequently mutated in NETs. Multiple clinical trials have been conducted to test the treatment effect of the mTOR inhibitor everolimus in GEP-NETs. In the RADIANT-3 trial, for patients with advanced P-NETs, everolimus treatment led to a significantly longer median PFS (11 mo vs 4 .6 mo in the control group; HR: 0 .35 )[94 ]. In the RADIANT-4 trial, patients with advanced nonfunctioning GI and lung NETs had a longer median PFS in the everolimus arm (11 movs3 .9 mo in the control group; HR: 0 .48 )[95 ]. Everolimus is approved for patients with advanced P-NETs and nonfunctioning GI NETs.
Immune checkpoint inhibitors and antibodies targeting programmed cell death protein-1 (PD-1 ),programmed cell death protein ligand-1 (PD-L1 ), or cytotoxic T lymphocyte-associated antigen 4 (CTLA-4 ), have demonstrated promising therapeutic responses in various types of cancers[96 ]. Based on the durable antitumor efficacy and favorable safety profile in patients with advanced metastatic Merkelcell carcinoma, a high-grade cutaneous NEC[97 ], immunotherapy has been proposed to be potentially effective for advanced NENs with microsatellite instability, high tumor burden, and/or mutational load[98 ]. Multiple clinical trials have been conducted to test the efficacy of immunotherapy in GEP-NENs.Currently, these studies only showed very limited therapeutic effects for GEP-NENs[99 -101 ].Interestingly, in a phase 1 b trial on toripalimab (an anti-PD-1 antibody) for patients with high-grade NENs, patients with PD-L1 expression greater than 10 % and/or high tumor mutational burden (TMB)had a better objective response rate (ORR) than low PD-L1 (< 10 %) (50 .0 % vs 10 .7 %, P = 0 .019 ) and low TMB patients (75 .0 % vs 16 .1 %, P = 0 .03 )[100 ]. Therefore, PD-L1 expression is a potential therapeutic and prognostic biomarker for GEP-NENs.
CONCLUSION
GEP-NENs are relatively rare tumors, although the incidence rates have been steadily increasing over the past three decades. GEP-NENs consist of a genetically heterogeneous group of tumors ranging from slow-growing, well-differentiated NETs to aggressive, poorly differentiated NECs. Great progress has been made toward understanding their unique molecular genetics and combating advanced disease through improved diagnostic tools and effective therapeutic regimens. A multidisciplinary and personalized treatment approach would be crucial to achieve optimal clinical outcomes for patients with GEPNENs.
FOOTNOTES
Author contributions:Yin F wrote and finalized the manuscript; Wu ZH critically reviewed the manuscript; Lai JP collected and analyzed the data, made the figures, and finalized the manuscript; all authors have approved the final manuscript.
Conflict-of-interest statement:The authors declare that they have no competing interests in this study.
Open-Access:This article is an open-access article that was selected by an in-house editor and fully peer-reviewed by external reviewers. It is distributed in accordance with the Creative Commons Attribution NonCommercial (CC BYNC 4 .0 ) license, which permits others to distribute, remix, adapt, build upon this work non-commercially, and license their derivative works on different terms, provided the original work is properly cited and the use is noncommercial. See: https://creativecommons.org/Licenses/by-nc/4 .0 /
Country/Territory of origin:United States
ORCID number:Feng Yin 0000 -0002 -8444 -1123 ; Zi-Hao Wu 0000 -0003 -4188 -3527 ; Jin-Ping Lai 0000 -0001 -5365 -2481 .
S-Editor:Fan JR
L-Editor:Wang TQ
P-Editor:Fan JR
杂志排行

World Journal of Gastroenterology的其它文章
- Current status and future of targeted peptide receptor radionuclide positron emission tomography imaging and therapy of gastroenteropancreatic-neuroendocrine tumors
- Biliary metal stents should be placed near the hilar duct in distal malignant biliary stricture patients
- Clinical outcomes of endoscopic papillectomy of ampullary adenoma: A multi-center study
- Gut homeostasis, injury, and healing: New therapeutic targets
- Sirtuin1 attenuates acute liver failure by reducing reactive oxygen species via hypoxia inducible factor 1 α
- Peroxisome proliferator-activated receptor-alpha activation and dipeptidyl peptidase-4 inhibition target dysbiosis to treat fatty liver in obese mice