Gut homeostasis, injury, and healing: New therapeutic targets
2022-06-11SemaOncelMarcBasson
Sema Oncel,Marc D Basson
Abstract The integrity of the gastrointestinal mucosa plays a crucial role in gut homeostasis, which depends upon the balance between mucosal injury by destructive factors and healing via protective factors. The persistence of noxious agents such as acid, pepsin, nonsteroidal anti-inflammatory drugs, or Helicobacter pylori breaks down the mucosal barrier and injury occurs. Depending upon the size and site of the wound, it is healed by complex and overlapping processes involving membrane resealing, cell spreading, purse-string contraction,restitution, differentiation, angiogenesis, and vasculogenesis, each modulated by extracellular regulators. Unfortunately, the gut does not always heal, leading to such pathology as peptic ulcers or inflammatory bowel disease. Currently available therapeutics such as proton pump inhibitors, histamine-2 receptor antagonists, sucralfate, 5 -aminosalicylate, antibiotics, corticosteroids, and immunosuppressants all attempt to minimize or reduce injury to the gastrointestinal tract. More recent studies have focused on improving mucosal defense or directly promoting mucosal repair. Many investigations have sought to enhance mucosal defense by stimulating mucus secretion, mucosal blood flow, or tight junction function. Conversely, new attempts to directly promote mucosal repair target proteins that modulate cytoskeleton dynamics such as tubulin, talin,Ehm2 , filamin-a, gelsolin, and flightless I or that proteins regulate focal adhesions dynamics such as focal adhesion kinase. This article summarizes the pathobiology of gastrointestinal mucosal healing and reviews potential new therapeutic targets.
Key Words: Intestine; Mucosa; Repair; Restitution; Sheet migration; Stomach ulcer
INTRODUCTION
Upon injury, gastrointestinal (GI) epithelial tissue is capable of renewing itself within hours to months by replacing damaged or dead cells, depending on the site and size of the wound. In order to appreciate potential new therapeutic targets, this review will first summarize the current understanding of the processes of mucosal healing and defense and describe their major extracellular regulators. Then,the importance of the quality of ulcer healing and novel approaches to promote such healing will be reviewed. This review focuses on mucosal injury and repair. Deeper injuries such as a deep ulcer,trauma, fistula, or surgical transection and anastomotic healing all require a complex interaction among endothelial cells, fibroblasts, and other cell types to reconstitute the submucosal and muscular layers of the bowel wall. This is beyond the scope of the current review but has been previously reviewed[1 -5 ].Angiogenesis is critical to these efforts, and requires a complex interaction between endothelial cells, the extracellular matrix, growth factors and cytokines, and other cell types[6 ,7 ].
PHYSIOLOGY OF MUCOSAL HEALING
The integrity of the gastrointestinal mucosa is crucial for gut homeostasis. The gut lining is continuously injured during normal gut function[8 ] by a variety of noxious luminal substances and abrasive interactions with luminal contents (Figure 1 A). However, there is normally an equilibrium between gut injury, mucosal healing, and diverse factors that protect the mucosa[5 ]. This equilibrium favors healing in a healthy state. Under normal physiological conditions, GI epithelial cells migrate from the base of the crypt to the villi, where their interaction with each other and the extracellular matrix (ECM) is disrupted leading to epithelial cell shedding (anoikis)[9 ] (Figure 1 B).
PROTECTIVE FACTORS FOR THE GASTROINTESTINAL MUCOSA
The gastrointestinal mucosa is protected at three levels: pre-epithelial, epithelial, and sub-epithelial defenses. Pre-epithelial protection, the first line of mucosal defense, is provided by the secretion of mucus, bicarbonate, phospholipids, prostaglandins, and trefoil peptides (Figure 1 A and B). These factors not only neutralize the acid but also inactivate pepsin at the gastric mucosal surface. In addition,phospholipids secreted into mucus contribute to the hydrophobicity of mucus and prevent backdiffusion of hydrogen ions[10 ]. Prostaglandins are abundant in gastric juice. They inhibit acid secretion and stimulate mucus and bicarbonate secretion[11 ]. Bicarbonate-rich mucus is secreted throughout the GI tract, by mucoid cells in the stomach and goblet cells in the intestines, creating a near-neutral pH at the epithelial surfaces in the GI tract, thereby protecting the GI mucosa against autodigestion by the gastric juice and other noxious agents in the lumen[12 ,13 ].
Intestinal epithelial cells consist of four important cell types (Figure 1 E and F). Enterocytes and colonocytes are most common in the surface epithelium. They are critical for the digestion and absorption of nutrients. Paneth cells are highly specialized cells located in small intestinal crypts. Paneth cells are essential for the secretion of antimicrobial peptides (AMP) such as defensins. These AMPs modulate the composition of the small intestinal microbiota. Goblet cells produce various types of mucin and are found throughout the GI tract. Similarly, enteroendocrine cells are scattered along with the epithelial cells of the GI tract. They produce and secrete more than 20 different hormones in response to nutrients in the lumen that regulate hunger, appetite, and satiety[14 ]. Intestinal epithelial stem cells (IESCs) are crucial to maintaining intestinal epithelial function and homeostasis in both the small intestine and large intestine. IESCs divide for self-renewal and generate progenitor cells that undergo differentiation into enterocytes, colonocytes, Paneth cells, goblets cells, and enteroendocrine cells[15 ].

Figure 1 Normal gastrointestinal homeostasis, injury, and healing. A: Structure of gastric epithelium in healthy, injured, and repaired states. A healthy gastric barrier is essential to maintain gastric homeostasis. In a healthy state, there is an equilibrium between gastric injury and mucosal healing. An excess of destructive factors such as acid, pepsin, nonsteroidal anti-inflammatory drugs (NSAIDs), and H. pylori leads to gastric barrier disruption. These noxious agents then diffuse deeper into the mucosa and create wounds. Epithelial cells at the edge of the injury redifferentiate to a migratory phenotype and collectively migrate as a sheet to close the wound. After successful restitution, the migrated cells redifferentiate to more specialized phenotypes. B: A diagram depicting the structure and cell types of gastric epithelium. C: In the injured state, epithelial cells at the edge of the wound spread and redifferentiate to a migratory phenotype, losing their classical apical brush border and assuming a more squamous morphology. Then, they migrate as a sheet to cover the injured area, with cells at the front of the migrating sheet transmitting traction forces to cells farther back via cell-cell contacts. Epithelial cells behind these migrating cells subsequently proliferate to provide more cells to fully cover larger wounds. D: Cells that have migrated across the defect may themselves then proliferate once the barrier has been reformed. In addition, following migration and proliferation, the migrated cells redifferentiate back to more specialized phenotypes. E: Structure of small intestinal epithelium in healthy and injured states. F: Structure of large intestinal epithelium in healthy and injured states. A healthy intestinal barrier is essential to maintain intestinal homeostasis. In the healthy state, there is an equilibrium between intestinal injury and mucosal healing. An excess of destructive factors such as NSAIDs, inflammation, bile acid, and toxic luminal substances leads to intestinal barrier disruption. These noxious agents then diffuse deeper into the mucosa and create wounds. Epithelial cells at the edge of the injury follow the processes described in the figure legends for in Figure 1 C and D. IESC: Intestinal epithelial stem cells; EEC: Enteroendocrine cells; GC: Goblet cells; NSAIDs: Nonsteroidal anti-inflammatory drugs; H. pylori: Helicobacter pylori; PG: Prostaglandins; ECL cells: Enterochromaffin-like cells; PC: Paneth cells;IESC: Intestinal epithelial stem cells.
Epithelial protection represents the second line of defense. GI epithelial cells are connected to each otherviatight junctions and act as a physical barrier against acid or toxic luminal agents. In addition to stimulating mucus and bicarbonate secretion, prostaglandins reduce the permeability of the epithelium by closing the apical spaces between the epithelial cells, and thus decreasing the exposure of deeper layers along the GI tract to noxious agents by modulating these tight junctions[16 ]. Tight junction proteins are either transmembrane proteins such as occludin, claudins, and junction adhesion molecule proteins or cytoplasmic plaque proteins such as the zonula occludens proteins. The dysregulation of these proteinsviatoxin exposure or autoimmune diseases such as celiac disease may lead to disruption of gastrointestinal barrier function[17 ]. For example, ulcerative colitis may alter the intestinal barrier functionviachanging the phosphorylation of colonic claudins[18 ]. The architecture and function of tight junctions are slightly divergent between the different regions of the GI tract and also between different epithelial cells. For example, disruption of occludin alters intestinal barrier function whereas occludin disruption does not cause barrier dysfunction in the stomach[19 ]. Moreover, the expression of tight junction proteins varies even among the different epithelial cells. For instance, IESCs and Paneth cells have high occludin levels whereas claudin-1 , -2 , and -7 expression is elevated in Paneth cells, IESCs, and enterocytes, respectively[20 ].
The final mucosal defense is sub-epithelial protection through augmentation of mucosal blood flow.Vascular flow not only removes acid rather than allowing it to diffuse deeper into the mucosa but also supplies necessary nutrients and oxygen to the epithelial cells for energy-consuming processes such as ion transport and secretion. Gut epithelial cells undergo continuous dynamic self-renewal in response to the damage caused by destructive factors under normal physiological conditions[21 ,22 ].
DRIVERS OF MUCOSAL INJURY
Although the gut epithelium can maintain normal homeostasis in the presence of modest or transient exposure to injurious stimuli, high level or extensive interactions with noxious factors such as excessive secretion of gastric acid and pepsinogen, the substantial inflammation caused by inflammatory bowel disease, or toxic luminal contents including ethanol or medication such as non-steroidal anti-inflammatory drugs (NSAIDs) can unbalance the equilibrium between mucosal injury and healing(Figure 1 A).
Gastric juice includes mucus, hydrochloric acid (HCl), bicarbonate, pepsin, and intrinsic factor secreted by mucoid cells, parietal (oxyntic) cells, and chief (zymogenic) cells in the stomach (Figure 1 B).Helicobacter pylori (H. pylori)infection,a gram-negative bacterium responsible for 90 % of duodenal and gastric ulcers, impairs the bicarbonate secretion and promotes gastric acidity as well[23 ]. Such hyperacidity may injure the mucosa, causing gastritis, duodenitis, peptic ulcer disease (PUD), or gastroesophageal reflux disease (GERD)[24 ].
The gastrointestinal mucosa is subjected to numerous physical forces such as strain and pressure during both normal gut function and illness. For instance, luminal chyme, peristalsis contractions,rhythmic villous motility, and some pathological conditions such as inflammatory bowel disease (IBD)may adversely impact GI mucosal healing by increasing luminal pressure[25 -29 ]. Such pressure increases have been shown to inhibit mucosal healing, at least in mice, despite increased mucosal proliferation, and appear to act by inhibiting the cell motility required for restitution[28 ].
The balance between mucosal injury and healing may also be shifted by drugs such as NSAIDs,corticosteroids, bisphosphonates, potassium chloride, steroids, and fluorouracil[23 ,30 ,31 ]. In particular,many studies have documented that NSAIDs decrease mucus hydrophobicity as measured by contact angle goniometry whereas prostaglandins, gastroprotective compounds, increase the contact angle of gastric mucosa[32 ,33 ]. NSAIDs, the most commonly prescribed medications, increase the development of ulcers in the upper and lower GI tract by two distinct mechanisms (Figure 2 )[34 -37 ].
NSAIDs injure the upper GI mucosa mainly by cyclooxygenase (COX)-1 inhibition, resulting in a decrease in prostaglandins, mucus, and bicarbonate secretion. Moreover, NSAIDs also alter another important component of mucosal defense, the gastric microcirculatory system. Upon irritation, the gastric mucosa normally increases blood flow to remove any toxins, bacterial products, or backdiffusing acid. Impairment of this hyperemic reaction increases the vulnerability of gastric mucosa to damage[38 ]. Inhibition of prostaglandins, potent vasodilators, by NSAIDs leads to an increase in vascular tone and thus reduces gastric mucosal blood flow[39 ], consequently, increases ischemic tissue damage and exacerbating the mucosal injury[40 ]. NSAIDs may also induce local gastric mucosal injury independent of prostaglandin deficiency[41 ]. NSAIDs may lyse phospholipids from mucosal epithelial cells and may increase mucosal permeability, which then allows mucosal exposure to luminal aggressive factors such as bacteria and gastric acid[42 ].

Figure 2 Non-steroidal anti-inflammatory drugs induce mucosal injury in the upper and lower gastrointestinal tract by two distinct mechanisms. In addition to principal luminal aggressors such as acid, pepsin, Helicobacter pylori in the stomach and acid, bile, and pathogens in the small intestine, nonsteroidal anti-inflammatory drugs (NSAIDs) increase mucosal damage in both upper and lower GI by two different mechanisms. In the stomach, the inhibition of COX-1 by NSAIDs reduces prostaglandin secretion which in turn reduces mucus and bicarbonate secretion and increases acid secretion, resulting in increased permeability and eventually mucosal damage. In the small intestine, NSAIDs bind to bile in the enterohepatic circulation. This potentiates the mucosal damage caused by bile. NSAIDs also increase mucosal damage in the small intestine by altering the gut microbiota. The NSAID-associated increase in enteric gramnegative bacteria appears to contribute to intestinal lesions by increasing inflammation. NSAIDs: Nonsteroidal anti-inflammatory drugs.
The molecular and cellular mechanisms of NSAID-induced lower GI mucosal injury are clearly distinct from NSAID-induced upper GI injury[42 ,43 ]. As in the stomach, NSAIDs may inhibit COX-1 and contribute to mucosal damage. However, unlike gastric injury, the bile acid and intestinal microbiota play a crucial role in the pathophysiology of NSAID-induced intestinal injury[42 ,44 ].NSAIDs and gut microbiota have complex and dynamic interactions. The gut microbiota can alter the efficacy and toxicity of NSAIDs either directly by biotransforming them into metabolites or indirectly by altering the host metabolism (e.g., interfering with hepatic function)[45 ]. On the other hand, NSAIDs themselves can directly change the composition and function of the gut microbiota or indirectly by altering the physiological functions of the host[45 ]. For instance, NSAIDs alter the intestinal microbiome by increasing the total number of bacteria and the proportion of gram-negative bacteria, which seems to be linked to the activation of toll-like receptor (TLR) 4 that increases inflammation and contributes to an intestinal injury[46 -48 ].
NSAIDs make complexes with bile acids by glucuronidation in the liver. This interaction alters the stability and structure of bile acids and potentiates bile acid toxicity in the lower GI tract[42 ]. These NSAID-bile acid complexes are secreted into the duodenum and subsequently reabsorbed back in the ileumviathe enterohepatic circulation. Within the intestinal lumen, particularly, in the colon,conjugated primary bile acids are deconjugated into more toxic secondary bile acids, mainly by the gram-positive bacteria[49 ]. There is crosstalk between the microbiome and the bile acids because bile acids can control the composition of the intestinal microbiome, which in turn regulates the composition and size of the bile acid pool[50 ,51 ]. Alteration in the colonic microbiota may cause a shift towards to generation of more toxic secondary bile acids, which eventually increase intestinal permeability, particularly in the colon, bacterial translocation, and mucosal inflammation[52 -54 ].
NSAID-induced ulcers are traditionally treated with proton pump inhibitors (PPIs) or histamine-2 receptor antagonists (H2 -antagonists)[55 ,56 ], which permit ulcer healing by reducing gastric acid secretion without directly affecting mucosal restitution[5 ,57 ,58 ]. Although PPIs have historically been co-prescribed with NSAIDs to ameliorate gastroduodenal injury and are used to treat NSAID injury,such use may increase the risk of a different problem. There is no evidence that gastric acid plays a key role in the pathogenesis of NSAID-induced lower GI[42 ,59 ]. PPIs may worsen NSAID-induced enteropathy by increasing gastric pH and thus changing the enteric microbiome by increasing the number of gram-negative bacteria[35 ,42 ,60 -62 ]. Thus, even though PPIs are still recommended to treat upper GI ulcers, their prophylactic use with NSAIDs to prevent upper GI injury is no longer recommended unless the patient has a moderate to high risk of peptic ulcer disease[62 ,63 ]. Similar concerns are likely to exist for H-2 blockers.
Inflammatory bowel disease is a broad term to describe disorders including Crohn’s disease (CD) and ulcerative colitis (UC) that are characterized by excessive activation of the mucosal immune system to normal microflora. This causes chronic inflammation and damages the gut mucosa[64 ,65 ]. Since the etiology of IBD is still unclear, the primary goal of treatment is centered on the elimination of inflammation with medical therapies such as 5 -aminosalicylate, antibiotics, corticosteroids, immunosuppressants, and biological therapy[66 -68 ]. Management of IBD with targeted therapies has been discussed in detail in a recent review[68 ]. However, none of these therapies is perfect, and even if patients achieve symptomatic remission, maintaining that remission can be challenging[69 ]. Recent evidence highlights the importance of mucosal healing over and above symptomatic remission in the quality of life and long-term prognosis of IBD patients[70 -72 ].
MUCOSAL HEALING PROCESSES
Once an injury has occurred, diverse processes such as redifferentiation to a migratory phenotype[73 -75 ], migration, proliferation, and eventual redifferentiation back to more specialized cells after healing are all regulated by various factors including growth factors, cytokines, physical forces, and the extracellular matrix itself. These coordinate healing of the injury (Figure 1 ). At the subcellular level,wounding of the apical plasma membrane is common in the epithelial cells of the intact, normal functioning stomach and intestinesin vivoafter mechanical and chemical stressors[8 ,76 ,77 ].Since maintenance of plasma membrane integrity is essential for cell viability, the wounded cell rapidly repairs the injury to restore internal homeostasis and prevent cell death. Plasma repair processes such as tension reduction, budding, patch, endocytosis, and exocytosis may be triggered by the toxic level of Ca2+ influx through the plasma membrane wound to then reseal the injured plasma membrane[78 ,79 ].
Relatively small or superficial multicellular mucosal injury undergoes complex wound healing processes that quickly reconstitute the mucosal barrier, depending on the size and depth of the injury.Small wounds, less than eight cells in size, may close by the spreading of neighboring cells and formation of new cell-cell contacts[80 ,81 ] or by purse-string wound closure, which involves the formation of a multicellular actin cable purse string around the wound, with actin cables that parallel the wound edge. This then contracts, pulling the adjacent cells together[5 ,82 ].
Mucosal injury involving more than eight cells is generally too large for purse-string wound closure.This then requires restitutive epithelial sheet migration to close the injury. Depending on the size and depth of these larger wounds, wound closure will require a longer healing time and may require one or more complex overlapping processes such as differentiation, proliferation, and angiogenesis for wound healing[5 ,83 ,84 ].
Restitution requires a phenotypic redifferentiation. Although some authors describe the initial steps of this process as dedifferentiation, it is the firm opinion of the senior author that this should rather be considered a redifferentiation toward a migratory phenotype. The gut epithelium normally consists of a monostratified layer of differentiated epithelial cells. At the edge of a mucosal wound, epithelial cells change their phenotype from differentiated columnar enterocytes or gastric cells to a migratory phenotype. They lose their typical morphology and (for enterocytes and parietal cells) their microvilli[85 ], disassemble their apical specialized membrane components[86 ], flatten out and extend lamellipodia toward the defect. Such migrating cells adopt a squamous morphology with altered integrin[87 -89 ] and cytoskeletal organization[73 ,85 ] and specialized cell signaling pathways[90 -93 ] that adapt these cells toward motility (Figure 1 C)[73 ,85 ,94 -96 ] Moreover, it is worth noting that these signaling events are not only regulated by the activation of signaling proteins but also by the distribution and the amount of the signaling proteins within the migrating cells. For instance, both the actual amount of total focal adhesion kinase (FAK) and the amount of active FAK decrease while the ratio of activated to total FAK increases both in vitro[94 ] and in vivo[92 ] as the epithelial cells shift to the migratory phenotype[73 ]. Similarly, both paxillin protein and tyrosine-phosphorylated paxillin decrease in migrating cells compare to static cells[94 ]. (Paxillin is an adapter protein critical to focal adhesion complex assembly and disassembly in response to various stimuli.)[97 -100 ]. Total p38 , ERK1 , and ERK2 proteins do not show differences between migrating and static cells[94 ]. However, phosphorylated p38 increases, and phosphorylated ERK1 and ERK2 decreases in motile cells compared with nonmigrating cells[94 ].
Furthermore, the distribution of these signaling proteins also changes in migratory phenotype. In confluent cells, FAK localizes mainly in a perinuclear pattern while FAK appears explicitly at the cell borders contacting other cells in motile cells, with FAK immunoreactivity decreasing toward the migrating lamellipodia that face the wound edge[94 ]. In contrast to FAK, paxillin is localized at the lamellipodial edges in migrating cells[94 ]. The difference is more than semantic because considering these migratory cells as a specialized phenotype opens up the possibility for therapy to modulate that phenotype and thereby promote mucosal healing.
The transverse actin cables that drive purse-string closure for smaller wounds line up parallel to the wound edge at the migrating front, connected by cell-cell contacts, and unite the migrating front, so that these redifferentiated cells collectively migrate as a sheet, a.k.a., restitution, to close the wound(Figure 1 C)[101 -103 ]. Slightly deeper wounds that injure the basement membrane expose the cells to the interstitial extracellular matrix. While the basement membrane is predominantly laminin and type IV collagen, the deeper interstitial matrix is rich in type I collagen, across which the cells may migrate more rapidly[104 ,105 ].
After the closure of the wound by successful restitution, the migrating cells must redifferentiate back to the more specialized phenotypes required for the normal biology of the mucosa (Figure 1 D)[102 ].Tarnawskiet al[106 ] have demonstrated the critical relationship between defective redifferentiation of these migratory cells and subsequent ulcer recurrence. This will be considered in more detail below.
If the wound surface area is extensive, restitution will likely be insufficient to seal the wound. In this situation, epithelial cell proliferation increases behind the migrating cells to create a larger pool of epithelial cells that can then migrate across and cover the defect (Figure 1 C)[107 ]. However, if the wound extends into deeper layers such as the submucosa and muscularis, these must also be reconstructed for healing by processes beyond the scope of this review. In particular, the reconstitution of nutrient vessels in the submucosa is critical for mucosal wound healing because these provide oxygen and nutrients to the mucosa and remove waste products from the wound site[108 ,109 ]. This neovascularization can occur by two distinct processes called angiogenesis and vasculogenesis[110 -114 ].Angiogenesis refers to the process where new blood vessels are formed from preexisting blood vessels from the wound’s adjacent vasculature by sprouting and forming tube-like structures and networks.Vasculogenesis is thede novoformation of new blood vessels from the differentiation of bone marrowderived progenitor stem cells.
RESTITUTION AND QUALITY OF ULCER HEALING AS THE SINE QUA NON FOR WOUND HALING
GI ulcers have traditionally been assessed in clinical settings by a superficial visual endoscopic examination that cannot assess the histological and ultrastructural characteristics of the mucosa or deeper layers. Ulcer recurrence is, unfortunately, common, with rates exceeding 60 % if the underlying problem has not been successfully addressed[23 ]. Recurrence of GI ulcers may be related to many factors including gastric acid secretion,H. pylori, NSAIDs, hormonal complications, size and depth of ulcers, anti-ulcer treatment, age, gender, comorbidity, alcohol consumption, and smoking[115 -118 ]. In 1991 , Tarnawski et al[119 ] drew attention to the relationship between recurrence of ulcers and ultrastructural abnormalities of deeper layers such as poor `redifferentiation, dilation of glands, reduced mucosal height, and disorganized microvascular network after ulcer healing and proposed the concept of the quality of ulcer healing (QOUH)[119 -121 ]. QOUH is defined as ideal ulcer healing, demonstrating flat ulcer scar, high functional restoration, and histological maturity of the regenerated tissue[115 ,122 ].Many patients treated with PPIs for GI ulcers still suffered from a recurrence of ulcers despite continuous anti-ulcer therapy[115 ,122 -124 ]. It appears that acid inhibition by PPIs or H2 -antagonists may be insufficient for successful high-quality gastroduodenal ulcer healing because low levels of prostaglandins and high levels of oxygen free radicals entail poor QOUH and thus potentiate ulcer recurrence[122 ,125 ,126 ].
Overall, cumulative data highlight the necessity of QOUH for successful and permanent ulcer healing and point out that contemporary treatments such as PPIs and H2-antagonists do not always provide such high-quality healing. Therefore, to improve QOUH and decrease the rate of recurrence of GI ulcers, new antiulcer drugs need to be developed to address this. Investigation of the endogenous biologic regulation of mucosal healing, suggests new therapeutic targets, both extracellular and intracellular.
REGULATORS OF MUCOSAL HEALING AND POTENTIAL NEW THERAPEUTIC TARGETS
Supplementing available therapeutic modalities that attempt to minimize or reduce injury, investigators have more recently focused on enhancing mucosal defense or promoting mucosal repair. Both mucosal defense and mucosal healing processes such as restitution, proliferation, angiogenesis, and vasculogenesis can be influenced by acid secretagogues, growth factors, trefoil peptides, cytokines, angiogenic factors, luminal nutrients, and the gastrointestinal microbiota[5 ,25 ,127 ]. In addition, physical forces like strain and pressure, engendered by peristalsis, villous motility, and interaction with luminal contents can influence intestinal epithelial migration and proliferation in a complex manner influenced by the deposition of fibronectin at the site of injury[25 ,128 ,129 ].
Acid secretagogues
Under physiological conditions, the stomach protects itself against various forms of endogenous and exogenous injury, primarily by gastric acid. Gastroprotective mechanisms could be triggered by acid secretagogues such as gastrin, histamine, and thyrotropin-releasing hormone (TRH)[130 -132 ].Pentagastrin, synthetic gastrin, stimulates gastroprotection in acidified aspirin-induced gastric injury in rats, likely through the activation of histamine-2 receptors, since this is abolished by ranitidine[133 ].However, exogenous gastrin protects the rat gastric mucosa against ethanol-induced lesions but not against aspirin-induced gastric damage in rats[134 ]. Several studies have shown that exogenous histamine-stimulated acid secretion also exerts a protective effect on the gastric mucosa against erosions induced by exogenous HCl in rabbits and frogs by stimulating a greater alkaline tide[135 ,136 ]. The central vagal activation by intracisternal injection of the thyrotropin-releasing hormone analog RX77368 enhances mucosal resistance as well by stimulating mucosal blood flowviaprostaglandin-independent manner which eventually results in the removal of diffused acid from the subepithelial interstitial space[137 ]. In addition, RX77368 increases the thickness of the mucus gelviaprostaglandin-dependent manner which slows down the acidification of surface cells[137 ]. The potential therapeutic adaptation of molecules like RX77368 and other acid secretagogues awaits the further exploration of the disparities between results depending on how the ulcers are induced, as well as challenges with their pharmacologic delivery.
Growth factors, trefoil peptides, and cytokines
Growth factors have diverse pathophysiologic effects, including cytoprotection against destructive agents, epithelial wound healing in response to injury[102 ,138 -140 ], and angiogenesis[141 -144 ].Epidermal growth factor (EGF) and transforming growth factor TGF-α are structurally related but different polypeptide growth factors[145 ]. They both bind to the same cell-surface EGF/TGF-α -receptor and induce generally similar effects[145 ].
EGF may act in a cytoprotective fashion against mucosal injury by increasing secretion of mucus and bicarbonate[146 -148 ], enhancing blood flow[149 -151 ], or releasing other cytoprotective agents such as prostaglandins[152 ]. Pretreatment of the stomach[153 ,154 ], small intestine[146 ,155 ], and colon[149 ,150 ]tissues, bothin vivoandin vitro, with EGF decreases mucosal damage by various noxious agents. TGF-α is similarly cytoprotective against gastric injury by ethanol, acetic acid, or aspirin[156 ,157 ]. Pretreatment of Caco-2 cells with EGF prevents deoxycholate-induced cellular damage, at least in part, by changes in intracellular calcium content[158 ], suggesting that EGF exerts direct cytoprotective effects on the epithelium in addition to its effects on blood flow and mucus secretion. Furthermore, because this EGFinduced cytoprotection was observed following only 30 minutes of pretreatment (insufficient for proliferation), these results also suggest that this protection is independent of the mitogenic effects of EGF[158 ]. Consistent with this idea, adding EGF to the basal surface of rabbit primary gastric epithelial cell monolayers cultured on collagen-coated inserts enhances cytoprotection against apical surface acid by opening the plasma membrane calcium channels and increasing intracellular calcium[159 ].
In addition to their cytoprotective effects, EGF and TGF-α also promote mucosal healing after injury,stimulating both cell motility and cell proliferation[104 ,140 ,154 ]. Indeed, part of the epithelial mucosal shift to a phenotype adapted to wound healing may be an increase in sensitivity to these growth factors.A recent study demonstrated a 75 -fold increase in the number of cells expressing detectable EGFreceptors at the ulcer margin after gastric ulcer induction in rats[160 ]. Either parenteral or local submucosal intra-ulcer injection of EGF caused a comparable acceleration in the healing of acetic-acidinduced rat gastric ulcers, at least in part by increasing gastric blood flow, decreasing gastric acid secretion, and upregulating COX-2 expression[161 ]. This is in agreement with previous reports suggesting that COX-2 -influences mucosal healing by regulating both the hyperemic response and epithelial cell proliferation[162 ,163 ].
The trefoil peptides may also offer new opportunities for therapy because they are important both for mucosal defense[164 -166 ] (by increasing the viscoelasticity of mucus[167 ,168 ]) and mucosal repair[169 -171 ] (by influencing reepithelization[171 ] and inflammation[172 ]). The trefoil factor (TFF) family includes TFF1 (also called pS2 ) expressed in gastric surface mucous cells, TFF2 (also called a spasmolytic polypeptide or SP) produced by mucus-producing gastric mucous neck cells, antral gland cells, and duodenal Brunner’s glands, and TFF3 (also called intestinal trefoil factor or ITF), predominantly produced by goblet cells of the small and large intestine and found abundantly within the mucus. A trefoil domain consists of three loops created by disulfide bonds and all TFFs are comprised of two trefoil domains[173 ]. Trefoil peptides have been detected in different forms including monomers,dimers, and complexes with other molecules. This influences the strength of their association with mucin[173 ]. In particular, TFF dimers tightly interact with mucin, increasing the viscosity and elasticity of mucus in comparison to the effect of TFF monomers[167 ,174 ].
The TFFs are mostly distributed to the basolateral domain of gastric neck cells and parietal cells in the stomach, the Paneth cells in the small intestine, and the crypt cells in the colon[175 ]. TFF interactions and specific functions have been discussed in detail in a recent review[176 ]. A specific TFF receptor has not yet been described. However, some binding and functional studies propose potential TFF receptors that may influence epithelial restitution. TFFs have been reported to bind to transmembrane proteins such as the β1 integrin subunit, CRP-ductin, CXC chemokine receptor (CXCR) 4 , CXCR7 , proteinaseactivated receptor (PAR) 2 , PAR4 , leucine-rich repeat and Immunoglobin-like domain-containing protein (LINGO) 2 , LINGO3 , and EGFR[177 -181 ]. TFF3 enhances wound healing by activating EGFR and inducing MAPK[182 ] and PI3 K/Akt signaling pathways in vitro[183 ] whereas TFF2 directly activates CXCR4 and enhances the phosphorylation of ERK1 /2 and Akt in gastric epithelial cells[184 ].Indeed, the CXCR4 antagonist AMD3100 blocks TFF2 -dependent gastric epithelial repair[170 ]. TFFs,specifically TFF2 and TFF3 , regulate epithelial motilityviaintegrin-binding and activating focal adhesion kinase as well[175 ]. TFF2 also promotes cell migration via PAR4 [185 ], while TFF3 activates PAR2 [186 ]. Furthermore, TFF2 peptide may be required for optimum activity of EGFR and/or EGF signaling in the stomach because heparin-binding EGF and TGF-α do not induce EGFR activation in the stomachs ofTff2KO mice[177 ].
Oral administration of trefoil peptides, recombinant human SP, or rat ITF protects the gastric mucosa against ethanol or indomethacin-induced injury in a prostaglandin-independent manner[164 ]. Similarly,a more recent study has also shown that both parentally and topically applied trefoil peptides reduce ethanol-induced gastric damage, assessed by measurement of gastric mucosal Na+leakage and area of macroscopic injury in rats[187 ]. Complementing these results, transgenic mice that overexpress human TFF1 display increased resistance to indomethacin-induced small intestinal damage[188 ] whereas ITFdeficient mice are more prone to ulceration and hemorrhage after oral administration of dextran sulfate sodium (DSS)[189 ], suggesting that trefoil peptides play an important role in GI mucosal protection.There are likely to be several mechanisms by which the trefoil peptides promote mucosal healing. For instance, exogenous recombinant TFF2 increases epithelial wound healing by decreasing inflammation by negatively regulating IL-12 production from macrophages and dendritic cells[172 ] whereas exogenous TFF3 activates epithelial wound healing via the Na/H exchanger-2 [171 ] and accelerates gastric repairviaa mechanism that does not require cyclooxygenase activation[170 ].
TGF-β expression increases in affected mucosa from patients with IBD[190 ], and at the edge of human gastric and colonic ulcers[92 ]. Intravenous administration of recombinant bone morphogenetic protein(BMP)-7 , a subfamily of TGF-β superfamily, for five days significantly accelerates the healing of trinitrobenzene sulfonic acid (TNBS)-induced colitis in rats by decreasing the expression of pro-inflammatory cytokines (IL-6 , TNF-b, ICAM-1 )[191 ]. It should be noted that all of these growth factors and cytokines mentioned in this section interact in a complex fashion, and TGF-β potentiates many of them[127 ,192 ,193 ]. TGF-β also stimulates the synthesis of FAK, a key intracellular signal protein for cell motility and proliferation[92 ].
Basic fibroblast growth factor (bFGF) and hepatocyte growth factor (HGF) stimulate the healing of acetic acid-induced gastric lesions in rats similarly when administered intraperitoneally or by local submucosal injection at the ulcer site, suggesting that these growth factors also accelerate mucosal repair[161 ]. The healing of gastric ulcers by bFGF and HGF may involve enhancement of gastric blood blow around the ulcer, suppression of gastric acid secretion, and upregulation of COX-2 expression[161 ].
When the mucus barrier fails due to overexposure to the noxious agents, acid-back diffusion occurs.In healthy mucosa, increased blood flow response rapidly increases the circulation of pH neutral or slightly alkaline blood through the mucosa to neutralize the diffused acid[103 ]. Moreover, new vasculature is needed to perfuse and support the newly forming tissue. Therefore, wounds deeper than the epithelial layer also require the formation of new blood vessels in granulation tissue for mucosal healing. Like restitution and proliferation, neovascularization is also modulated by growth factors.Vascular endothelial growth factor (VEGF), platelet-derived growth factor (PDGF), nerve growth factor(NGF), fibroblast growth factor (FGF), and angiopoietin-1 (Ang1 ) are essential for blood vessel regenerationviaangiogenesis and vasculogenesis following mucosal damage[141 -144 ] and can therefore facilitate deep wound healing[112 ,193 -195 ]. VEGF, the most potent angiogenic factor, is an indispensable regulator of angiogenesis, making it potentially an ideal candidate to induce angiogenesis/vasculogenesis in mucosal healing. Since VEGF is digested in the lumen by proteolytic enzymes,Joneset al[141 ] used a single injection of a nonviral naked DNA plasmid encoding VEGF and Ang1 directly into the injured area to reduce such deleterious effects. They demonstrated that local gene therapy with a combination of VEGF and Ang1 cDNAs increases gastric ulcer healing and generates more mature vessels and a more complete epithelial structure in acetic acid-induced gastric injury in rats, suggesting that a combination of growth factors may have better therapeutic potency than the use of any individual factor[141 ]. Similarly, local gene therapy with serum response factor (SRF) accelerates ulcer healing as well as muscle restoration in acetic acid-induced gastric ulcers in rats[196 ]. A recent study indicated that angiogenesis and vasculogenesis go hand in hand while forming new vessels in granulation tissue[197 ]. Delivering such naked genes to a damaged site at endoscopy may be a promising tool to treat such ulcers by increasing the bioavailability of essential GFs.
Cytokines are also involved in the regulation of mucosal barrier function at multiple levels including mucosal homeostasis and inflammation[198 ]. Proinflammatory cytokines such as tumor necrosis factor(TNF) and IL-13 are upregulated in the inflamed mucosa of IBD patients. Anti-TNF therapy promotes mucosal healing in many patients, but not all patients respond to anti-TNF therapy. Many investigators have therefore focused on other cytokines to improve mucosal barrier function. Unfortunately, many of these attempts have ended disappointingly. For instance, experimental colitis in mice is controlled by using ROR-gamma null Th17 cells, which cannot produce IL17 A/F and not induce colitis[199 ].However, an anti-IL17 A monoclonal antibody not only failed to improve CD in clinical trials but actually aggravated adverse symptoms[200 ]. IL-13 has seemed similarly promising in pre-clinical studies[201 -204 ], but a trial of IL-13 blockade in UC failed[205 ]. Such failures of blockade of pro-inflammatory cytokines have recently prompted attempts to use anti-inflammatory IL-10 family cytokines to promote colonic mucosal healing. IL-10 is a major anti-inflammatory cytokine that targets hematopoietic cells in various autoimmune diseases[206 ]. Gene therapy with a single intravenous injection of an adenoviral vector encoding IL-10 (AdvmuIL-10 ) diminishes TNBS-induced colitis in mice, decreasing histological injury scores, weight loss, stool markers of inflammation (IL-1 β and TNFR-II), and serum amyloid protein in comparison to empty cassette virus (Adv0 ) or PBS treated mice in TNBS-induced colitis model[207 ]. Gelatin microspheres containing IL-10 have been developed to increase local bioavailability as a sustained release preparation[208 ]. Gelatin-microsphere-IL-10 treatment remarkably decreases colonic inflammation in IL-10 (-/-) mice compared to treatment with IL-10 alone treatment, at least in part by decreasing IL-12 mRNA expression and down-regulating CD40 expression in macrophage-1 positive cells[208 ]. However, recombinant IL-10 did not improve clinical symptoms in Crohn’s disease[209 ]. Such disappointing results raise the possibility that manipulating a single cytokine may have unpredictable results because of its effects on the web of compensatory pro-inflammatory and anti-inflammatory cytokine pathways in the inflamed mucosa[198 ].
One cytokine that may be promising is IL-22 . Even though it belongs to the IL-10 family of cytokines,IL-22 is unlike IL-10 in that it targets non-hematopoietic epithelial cells. IL-22 is produced by, apart from the adaptive T cell, innate cells including innate lymphoid cells (ILCs), specifically ILC3 cells in the GI tract. IL-22 has dual roles in inflammation. It can act as a protective (anti-inflammatory) cytokine or a pathological (pro-inflammatory) cytokine[210 ]. IL-22 influences various tissue epithelial functions such as inflammation[211 ,212 ], barrier integrity, regeneration, wound healing[213 -215 ], and host defense against pathogens[216 ,217 ]. Beneficial effects of IL-22 have been demonstrated in various murine colitis models[210 ,218 ]. However, IL-22 actually appears to worsen the anti-CD40 -induced colitis model, in that neutralization of IL-22 reduces the weight loss and colitis scores caused by the anti-CD40 injection and administration of IL-22 then recreates the colitis[219 ]. Thus, although most animal studies raise the possibility that recombinant human (rh) IL-22 might be a promising therapy for IBD, it remains unclear which effect will be seen in human disease. However, as for other cytokines, the short half-life of rhIL-22 (less than 2 h) limits its clinical applications. Several groups have sought to overcome this obstacle by engineering recombinant fusion proteins with a half-life of 1 -2 wk to improve the cytokine’s pharmacokinetic properties. Currently, seven IL-22 clinical trials have been investigated for different indications. UTTR1147 A is a human IL-22 fusion protein that links the human IL-22 with the Fc portion of human immunoglobulin (Ig) G4 , which is prepared for IBD studies[220 ]. Extensivein vitroandin vivostudies suggest that UTTR1147 A decreases histologic colitis severity by a pathway involving STAT3 activation[220 ]. These pre-clinical studies demonstrate that UTTR1147 A is well tolerated and is not associated with increased inflammatory cytokines in mouse, rat, and monkey studies[220 ]. A randomized phase-I healthy volunteer study of UTTR1147 A demonstrated satisfactory safety and pharmacokinetic profile[221 ]. A phase-II open-label extension study to evaluate the long-term safety and tolerability of UTTR1147 A in patients with moderate to severe UC and CD continues with an estimated completion date in 2025 [222 ].
Therapeutic use of growth factors (except BMP-7 ) may be limited by their low protein stability[144 ].In addition, despite their beneficial effects on the GI tract, long-term or systemic use of any growth factors, trefoil peptides, or cytokines that stimulate cell proliferation, either for cytoprotection or for mucosal healing, may raise concerns about inducing hyperproliferative or dysplastic lesions and potential tumorigenesis. This remains an open issue for such mitogens.
Luminal nutrients and GI microbiota
Luminal nutrients and microbiota are also crucial for the maintenance and repair of the gut mucosa.Short-chain fatty acids (SCFAs) are produced by commensal microbiota, mostly by gram-positive anaerobic bacteria, and are essential for perpetuating intestinal health[223 ,224 ]. These SCFAs, especially butyrate, are a major energy source for enterocytes and support gut homeostasis[225 -227 ]. SCFAs may stimulate the differentiation of epithelial cells and their proliferation in vivo[228 -230 ], whereas they promote only differentiation in cell culture models but inhibit proliferation and migration[231 -235 ].Long known as an energy supply for colonocytes and enterocytes, SCFAs have attracted may also enhance gut barrier function. SCFAs decrease acid-back diffusion by dilating arterial walls and increasing blood flow in gut mucosa[236 ,237 ]. In addition, several studies have documented improved intestinal barrier function after SCFA supplementation[238 -240 ]. The SCFAs activate 5 ’ adenosine monophosphate (AMP) kinase and therefore promote tight junction assembly, which in turn enhances intestinal barrier function[241 ,242 ]. However, a recent clinical study found no evidence that butyrate monotherapy or a combination of three SCFAs offered any advantage over placebo in improving the disease activity index in ulcerative colitis patients receiving maintenance oral anti-inflammatory medication[243 ].
Amino acids such as arginine, histidine, and glutamine promote enterocyte proliferation and decrease mucosal permeability by regulating tight junction proteins[244 -247 ]. A recent study proposed that histidine and arginine play an important role in stimulating intestinal restitution, probably stimulating FAKviathe TGF-β/Smad2 signaling pathway[248 ]. Glutamine modulates the phenotype of gut epithelial cells by stimulating proliferation and decreasing differentiationin vitro[249 ]. Similarly, many studies have been shown that glutamine also promotes cell proliferation of intestinal epithelial cells in weanling mice[250 ] and weaning piglets[251 ], prevents mucosal injury, and regulates enterocyte restitution following acetic acid-induced intestinal injury in rats[252 ].
Biologically active phospholipids in milk, phosphatidylcholine (PC) and phosphatidic acid (PA), and their metabolites such as lysophosphatidic acid (LPA), all act to increase the barrier function of GI mucosa by increasing the hydrophobicity of the mucus[253 ]. This makes the tissue non-wettable[10 ] and provides mucosal protection against aspirin-induced gastric injury in mice[253 ]. Dietary essential omega-6 fatty acids can enhance the biosynthesis of prostaglandins and increase the GI mucosal barrier[254 ]. Milk fat globule-epidermal growth factor 8 (MFG-E8 ), a glycoprotein found in mammary epithelial cells but also produced by lamina propria macrophages, also plays a vital role in modulating enterocyte migration along the crypt-villus axis[255 ].
Extracellular matrix
Epithelial sheet migration during gut-healing requires crosstalk between focal adhesion (FA) complexes in the lamellipodium and the ECM. The extracellular matrix is an extremely dynamic meshwork comprised of proteins, glycosaminoglycans, and glycoconjugates. Its composition and organization differ between tissue types and with physiological and pathological conditions[256 ,257 ]. Besides its structural support, the ECM has a direct role in gastrointestinal wound healing by inducing extensive signaling cascades[258 -260 ]. Plasma and tissue fibronectin accumulating in deeper wounds also help to shift the cells to a phenotype that responds to repetitive deformation by increased motility rather than by classical differentiation[129 ,261 ]. ECM remodeling is performed by matrix proteinases such as matrix metalloproteinases (MMPs), lysyl oxidases, and heparanases[262 ]. The gelatinases, a subgroup of MMPs, consist of two proteinases gelatinase A (MMP-2 ) and gelatinase B (MMP-9 ). In particular, MMP-9 is upregulated in the inflamed intestinal mucosa of IBD patients[263 -267 ]. Furthermore, anti-gelatinase neutralizing antibodies have been reported effective in murine DSS-induced colitis[268 ]. However, a phase II, randomized, placebo-controlled study found that the MMP-9 inhibitor andecaliximab did not induce a significant symptomatic or endoscopic response in patients with active Crohn’s disease[269 ].This lack of efficacy in Crohn’s disease prompted the termination of another clinical trial of the same agent in active ulcerative colitis[270 ]. Thus, while modulation of matrix metalloproteinases remains an attractive target in IBD, further exploration of the science involved and the reasons for the failure of the clinical trial are needed.
Regulation of cytoskeleton
Epithelial restitution begins at the edge of the wound with the redifferentiation of epithelial cells.Reorganization of the actin cytoskeleton is controlled by the Rho family of GTPases including RhoA,Rac1 , and Cdc42 (Figure 1 C)[93 ,271 -273 ]. Epithelial cells then form protrusions called lamellipodia with new focal adhesions (FAs) at the leading edge of the motile cells. The migrating cell increases its contractile forces and disassembles focal adhesions at the rear edge allowing the entire cell to move forward[274 -276 ]. Cell-cell linkages[276 ] transmit this force to other cells behind the migrating front and stretch the epithelial layer across the wound as a sheet. This sheet migration is characteristic of epithelial cells and differs from the individual cell motility displayed by other cell types.
The cytoskeleton, a complex and dynamic network of actin filaments, microtubules, and intermediate filaments, is also an important factor in wound healing[277 ]. Epithelial restitution relies on the coordination of forward protrusions and retraction forces at the rear edge, which is orchestrated by the actin and microtubule cytoskeleton[278 ]. In the lamellipodium, the elongating actin filaments produce the driving forces for the protrusion while microtubules form a polarized network that permits organelle and protein transport throughout the cell during cell migration[279 ,280 ]. Intermediate filaments, however, are generally considered for the maintenance of the overall cell shape[280 ]. Alternatively, or in combination with therapy to reduce ongoing injury by improving mucus barrier function and promoting angiogenesis, one could consider attempting to directly stimulate restitution in order to accelerate barrier reconstitution. Thus, proteins that modulate cytoskeleton dynamics might be targeted for optimal wound repair. Fidgetin-like 2 (FL2 ), a microtubule-severing enzyme, regulates the organization of the microtubule cytoskeleton for faster and successful repair of murine wounds[281 ].Actin remodeling proteins such as talin[282 ], Ehm2 [283 ], filamin-a[284 ], gelsolin[285 ], and flightless I(Flii)[286 ] have also been identified as potential new targets for improved wound healing. Unlike other members of the gelsolin family, Flii inhibits actin polymerization and FA turnover, thus decreasing migration[286 ,287 ]. Flii neutralizing antibodies (FnAb) decreased wound area with a quicker rate of healing in porcine and murine models of wound healing, respectively[288 ,289 ].
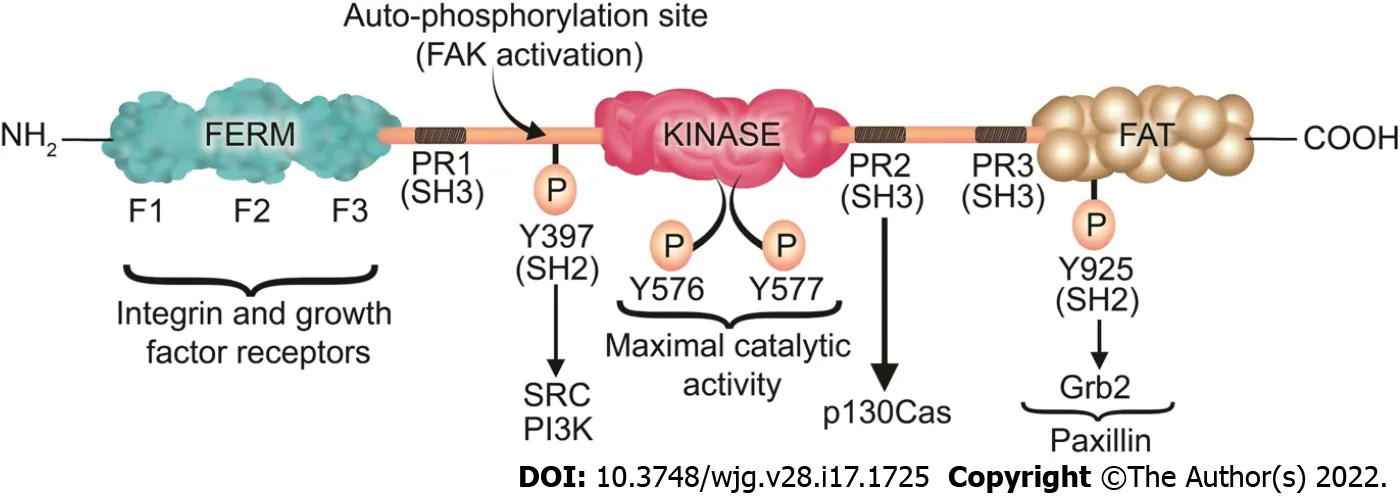
Figure 3 Focal adhesion kinase structure, phosphorylation sites, and its associated proteins. Focal adhesion kinase (FAK) contains an N-terminal band 4 .1 -ezrin-radixin-moesin (FERM) domain comprised of three lobes (F1 , F2 , and F3 ), a central kinase domain, a C-terminal FAT domain, and two linker domains with three PR regions that bind SH3 domain containing protein such as p130 Cas. Y397 is the site of the FAK autophosphorylation, crucial for FAK activation, which interacts with proteins containing the SH2 domain such as Src and PI3 K. Subsequently to the SH2 binding, Src binds to the PR1 SH3 domain (PXXP) and further phosphorylates the Y576 /577 sites on FAK, which are crucial for the maximal catalytic activity of FAK. Further FAK phosphorylation at Y925 creates a binding site for Grb2 . The phosphorylation of FAK-Y-925 and subsequent Grb2 binding disassociates paxillin from FAK, which results in FAK release from FAs, thus stimulating FA disassembly. The FERM domain regulates the interactions of FAK with growth factor receptors and integrins. The FAT domain recruits FAK to FAs by associating with paxillin. FERM: Band 4 .1 -ezrin-radixin-moesin; FAT: Focal adhesion targeting; PR: Proline-rich region; SH: Src homology; P: Phosphorylation.
Regulation of FAs
Cell migration, and consequently wound healing, depend critically on the dynamics of assembly and disassembly of FAs. The subunit composition of integrin receptors and the downstream signaling pathways may vary in different scenarios[290 -293 ]. Nevertheless, integrin binding to ECM triggers focal adhesion formation by recruiting many structural and signaling proteins including FAK, a non-receptor tyrosine kinase[294 -298 ]. FAK regulates FA dynamics both by recruiting other FA proteins such as Src to FA sites and by phosphorylating other signaling and adapter FA proteins such as paxillin and p130 Cas[299 -301 ]. FAK also influences the cytoskeletal remodeling essential for cell migration by regulating the Rho family of small GTPases such as Cdc42 , Rac1 , and RhoA[302 -305 ]. Inhibition of FAK inhibits cell migration[94 ,298 ].
Although FAK appears to activate cell motility and promote restitution, and FAK is indeed activated during cell motility, levels of both activated FAK and total FAK protein (including both active and inactive FAK) actually decrease in migrating GI epithelial cellsin vitroand at the edge of human gastric and colonic ulcersin vivoeven though the proportion of activated FAK increases (at leastin vitro)[92 ,94 ]. This reflects decreased FAK synthesis in cells that have adopted the migratory phenotype[306 ]. This apparently paradoxical reduction in this important protein makes FAK an attractive target for possible therapeutic intervention to promote mucosal healing.
FAK is a 125 kDa protein comprised of an N-terminal FERM (band 4 .1 -ezrin-radixin-moesin) domain,a central kinase domain, three proline-rich regions that are binding sites for Src homology 3 (SH3 )domain-containing proteins, and a C-terminal focal adhesion targeting (FAT) domain (Figure 3 ).
The FAT domain consists of a four-helix bundle[307 ] and is critical for targeting FAK to FAsviabinding to paxillin[308 ]. In an inactive (autoinhibited) state there is an interaction between the FERM and kinase domains which prevents FAK autophosphorylation at Y397 [309 ]. Upon competitive binding of candidate activating proteins such as the cytoplasmic regions of β-integrins or growth factor receptors on the F2 domain of FERM, the autoinhibited conformation of FAK is disassembled[310 ]. This conformational change allows Y397 phosphorylation, a key event in FAK activation[311 ]. In a subsequent step,Src is recruited and activatedviaSH2 binding to pY397 and SH3 binding to the PxxP sequence in the linker region, an essential step in promoting cell migration[311 ]. Then, Src phosphorylates the activation loop residues Y576 and Y577 of FAK and it acquires full catalytic activity after phosphorylation of the activation loop[312 ]. Phosphorylation of FAK at tyrosine 925 residue creates an SH2 binding site for the growth factor receptor-bound protein 2 (Grb2 ), adaptor protein[313 ]. The Grb2 binding site at FAK-Y-925 overlaps with one of the paxillin binding sites in the FAT domain of FAK[313 ]. The binding of Grb2 disassociates paxillin from FAK and potentiates the release of FAK from FAs[313 ]. On the other hand,paxillin acts as a scaffold protein for ERK signaling[305 ]. Subsequently, ERK may modulate FA turnover by further phosphorylating paxillin[305 ]. Therefore, Paxillin and Grb2 are critical FA proteins that interact with FAK and play an important role in FA turnover[97 ,313 ].

Figure 4 Focal adhesion kinase plays a crucial role in several signaling pathways that promote migration, proliferation, and survival. Upon its activation, focal adhesion kinase (FAK) directly binds PI3 K, leading to the activation of Akt. Activated Akt then stimulates numerous cellular functions including cell survival, proliferation, and migration via various signaling cascades depending on the cell type and species. In addition, FAK may recruit Grb2 and subsequently activate the Ras/Raf/MAPK pathway, enhancing cell proliferation and motility. Finally, FAK may directly bind to paxillin and p130 Cas, promoting lamellipodium formation, and thus migration via Rac GTPase activation.
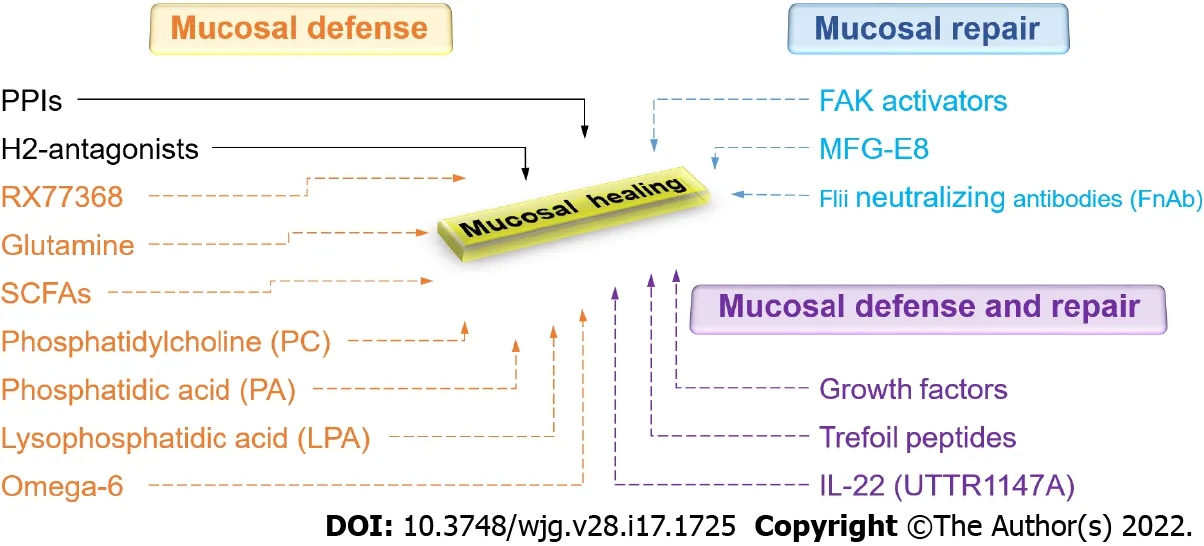
Figure 5 Current and promising new therapeutic approaches to gastrointestinal mucosal healing. Green represents currently available drugs.Red represents promising new therapeutic approaches that increase mucosal defense. Blue represents promising new therapeutic approaches that promote mucosal repair. Purple represents promising new therapeutic approaches that stimulate both mucosal defense and repair. PPIs: Proton pump inhibitors; H2 -antagonists:Histamine-2 receptor antagonists; RX77368 : The thyrotropin-releasing hormone analog; SCFAs: Short-chain fatty acids; FAK: Focal adhesion kinase; MFG-E8 : Milk fat globule-epidermal growth factor 8 ; Flii: Flightless I.
FAK has both a structural role as a scaffold for protein-protein interactions and a kinase function that phosphorylates many substrates in diverse signaling events[314 ,315 ]. Its non-kinase scaffolding function allows several different proteins to bind its N-terminal FERM domain and C-terminal FAT domain,tethering them into complexes (Figure 3 ). For instance, FAK may regulate cell migration serving as a scaffold for Src phosphorylation of p130 Cas[316 ] in FAs. Similarly, nuclear FAK may promote cell survival functioning as a scaffold to stabilize p53 -Mdm2 complexes, promoting p53 ubiquitination and proteasomal degradation[317 ]. On the other hand, in its kinase signaling capacity, FAK triggers many downstream signals including the Ras/Raf/MAPK[97 ,296 ,318 -320 ], p130 Cas-Crk[321 -324 ], and phosphatidylinositol 3 -kinase (PI3 K)-Akt pathways[317 ], which in turn coordinate to regulate cell proliferation, migration, and survival (Figure 4 )[313 ,325 ].
Recent evidence suggests that direct modulation of FAK activity is possible, practical, and effectiveviasmall molecule FAK activators[326 ]. A novel small molecule with drug-like properties,ZINC40099027 (ZN27 ), that mimics the FERM domain of FAK has been identified from the ZINC database and activates FAK in human intestinal epithelial cells without activating Pyk2 , the closest paralogue of FAK, or Src, another canonical nonreceptor tyrosine kinase within focal adhesions[327 ].Indeed, ZN27 directly activates both full-length 125 kDa and its 35 kDa kinase domain, increasing the maximal activity (Vmax) of FAK, suggesting that ZN27 is a highly potent and selective activator acting allosterically on the 35 kDa FAK kinase domain[328 ]. ZN27 not only activates FAK but also stimulates intestinal epithelial migrationin vitroand mucosal healing in mice after ischemic injury or injury by indomethacin[327 ]. ZN27 also activates FAK in gastric epithelial cells and promotes gastric mucosal healing in mice subjected to chronic ongoing injury by aspirin[58 ]. Structure-activity-relationship studies have developed a library of novel FAK activators based on ZN27 , that have drug-like properties,activate FAK, and stimulate epithelial sheet migration in vitro[329 ]. At least one such molecule (dubbed compound 3 ) demonstrates reasonable drug-like properties based onin vitro, in vivo, andin silicoresults with no obvious toxicity[329 ]. Further development of this lead molecule may offer the potential for a new therapeutic approach to actually stimulate mucosal healing by activating FAK.
CONCLUSION
Given the enormous impact of GI mucosal healing on human health, there is certainly a need to expand therapeutic options in this regard. A new understanding of the biology of mucosal healing suggests several different possibilities (Figure 5 ). These include FAK activators, UTTR1147 A, endoscopic gene therapy for angiogenic growth factors, mucus barrier enhancementviathe thyrotropin-releasing hormone analog RX77368 or trefoil peptides, enhanced energy for the mucosa with butyrate, and attempts to increase the regenerative ability of the epithelium with growth factors, cytokines, or trefoil peptides. Future work will determine which of these potentially promising avenues will prove successful and will need to balance their effects against potential risks and issues, including bioavailability, mitogenicity, and tumorigenesis.
FOOTNOTES
Author contributions:Oncel S and Basson MD equally contributed to this manuscript with regards to the conception and structure of the manuscript in addition to the literature review; The final draft of the manuscript was read and approved by all the authors.
Conflict-of-interest statement:The senior author (Basson MD) is co-inventor on patents applied for by the University of North Dakota describing the use of small molecule FAK activators to promote mucosal healing. The authors have no other conflicts of interest.
Open-Access:This article is an open-access article that was selected by an in-house editor and fully peer-reviewed by external reviewers. It is distributed in accordance with the Creative Commons Attribution NonCommercial (CC BYNC 4 .0 ) license, which permits others to distribute, remix, adapt, build upon this work non-commercially, and license their derivative works on different terms, provided the original work is properly cited and the use is noncommercial. See: https://creativecommons.org/Licenses/by-nc/4 .0 /
Country/Territory of origin:United States
ORCID number:Sema Oncel 0000 -0001 -5551 -2271 ; Marc D Basson 0000 -0001 -9696 -2789 .
S-Editor:Gong ZM
L-Editor:A
P-Editor:Gong ZM
杂志排行

World Journal of Gastroenterology的其它文章
- Current status and future of targeted peptide receptor radionuclide positron emission tomography imaging and therapy of gastroenteropancreatic-neuroendocrine tumors
- Biliary metal stents should be placed near the hilar duct in distal malignant biliary stricture patients
- Clinical outcomes of endoscopic papillectomy of ampullary adenoma: A multi-center study
- New insights in diagnosis and treatment of gastroenteropancreatic neuroendocrine neoplasms
- Sirtuin1 attenuates acute liver failure by reducing reactive oxygen species via hypoxia inducible factor 1 α
- Peroxisome proliferator-activated receptor-alpha activation and dipeptidyl peptidase-4 inhibition target dysbiosis to treat fatty liver in obese mice