重组人生长激素治疗IHH 基因杂合突变1 例
2022-06-08郑佳琪姜丽红牛乐乐马晨刘戈力
郑佳琪,姜丽红,牛乐乐,马晨,刘戈力
(天津医科大学总医院儿科,天津300052)
儿童纵向骨生长取决于生长板软骨形成的速率,这是一个受内分泌和旁分泌因素调节的复杂过程。任何影响生长板软骨形成的调控基因突变都可能导致身材矮小。随着高通量测序等分子生物学技术的高速发展,许多导致身材矮小的生长板调控基因不断被发现。印度刺猬(Indian hedgehog,IHH)基因编码了一个生长板重要的旁分泌调节因子——印度刺猬蛋白,该蛋白在调节软骨细胞分化、关节发育与骨形成等方面发挥重要作用[1]。IHH 基因杂合突变可导致身材矮小伴A1 型短指畸形(brachydactyly type A1,BDA1;OMIM 112500)[2]。目前国内对该基因突变少有报道。本文回顾性分析1 例以身材矮小就诊的IHH 基因杂合突变患儿接受重组人生长激素(recombinant human growth hormone,rhGH)治疗的临床资料,并进行相关文献复习,为临床诊治此病提供理论依据。
1 临床资料
1.1 一般资料 患儿,男,2 岁8 个月,主因“身材矮小”于2021 年4 月就诊于天津医科大学总医院儿科门诊。患儿系G2P1,其母孕期行超声检查发现胎儿股骨、肱骨短小,35 周时股骨相当于30 周+2 d,肱骨相当于28 周+2 d。患儿为足月顺产,出生时无产伤及窒息史,出生身长不详,出生体重3.6 kg。母亲身高155 cm,父亲身高159.5 cm,患儿祖母身高<150 cm(图1),父母非近亲婚配,否认家族遗传病史。体格检查:身高87.2 cm(P3),体重12 kg(P3~10),指距81 cm,坐高53.5 cm,坐高/下身长=1.58(P50~97)。身材匀称,无特殊面容,皮肤无色素沉着,甲状腺无肿大,心肺腹查体无异常,睾丸容积约2 mL,阴茎长度约3 cm,手足小,手指短小,第5 指中节指骨短(图2)。辅助检查:血常规、电解质、肝肾功能、甲状腺功能未见异常。染色体46,XY。胰岛素样生长因子1(IGF-1)47.9 ng/mL(-1SD~M),胰岛素样生长因子结合蛋白3(IGF-BP3)3.23 μg/mL(+1SD~+2SD)。25 羟维生素D 36.22 nmol/L,提示维生素D 缺乏。生长激素激发试验:峰值10.8 μg/mL,不支持生长激素缺乏。影像学检查:左手正位片:骨龄2.3 岁(骨龄比年龄落后0.4 岁),远节指骨短粗,第五指中节指骨短且形态不规则,见图3。腰骶椎正侧位:未见异常。垂体磁共振:垂体体积小,高度约1.8 mm,信号未见异常,垂体柄居中,无增粗。
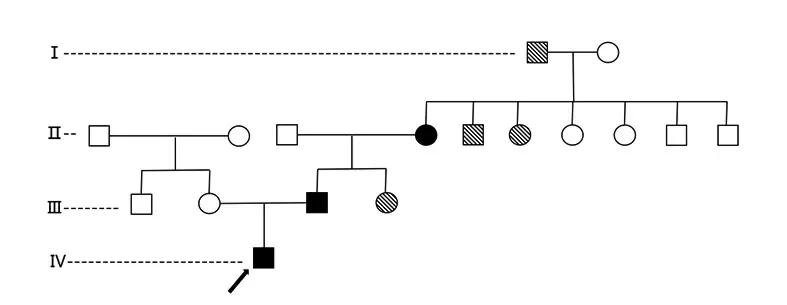
图1 家系图
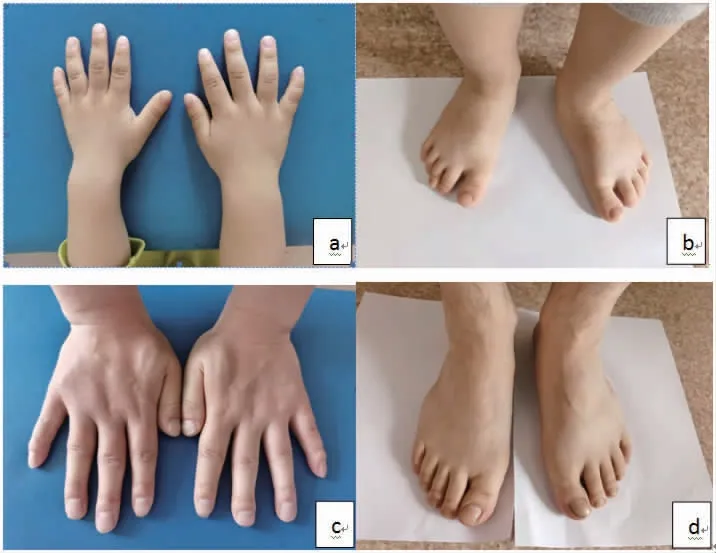
图2 患儿及患儿父亲手足照
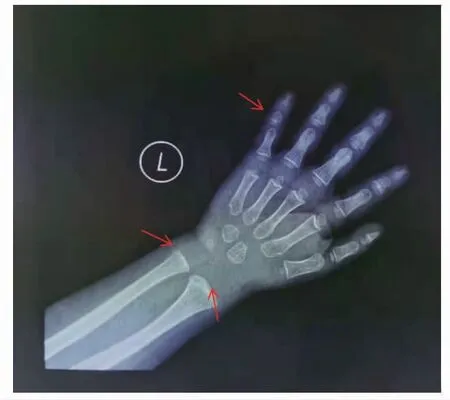
图3 患儿左手正位片
1.2 遗传学检查及生物信息学分析 患儿为家族性矮小(父亲身高<160 cm),且患儿与其父亲均有手足小、指趾短粗等表现,进一步完善遗传学检查:经医学伦理审核及患儿家属知情同意,完善全外显子组检测及Sanger 测序验证。结果提示IHH 基因有1 个杂合突变:c.174G>C(编码区第174 号核苷酸由鸟嘌呤变异为胞嘧啶),导致氨基酸改变p.E58D(第58 号氨基酸由谷氨酸变异为天冬氨酸),为错义突变。该位点位于Hedgehog 氨基末端信号域,在各物种中高度保守(图4a)。c.174G>C/p.E58D的功能预测如下(表1):在生物信息学蛋白功能综合性预测软件REVEL 预测结果为有害,SIFT、PolyPhen_2、MutationTaster 、GERP+预测结果分别为有害、有害、有害、有害。使用SWISS MODEL 软件预测IHH 野生型和突变体的3D 结构(图4b、4c)。结果表明:与野生型蛋白相比,突变蛋白中氢键位置发生改变,氢键位置由glu58-his187,变为asp58-ser189,可能导致IHH 蛋白空间构象改变。
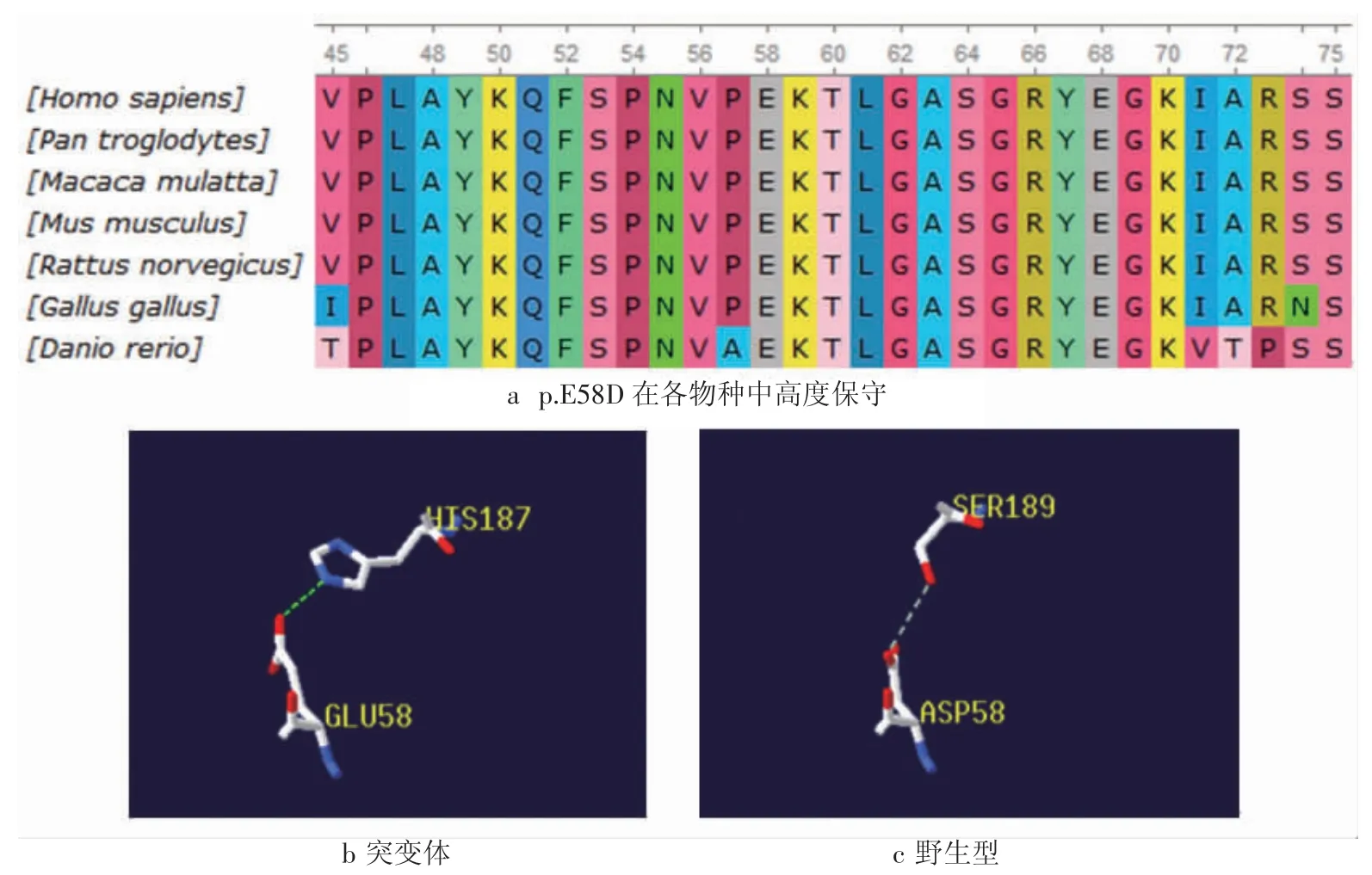
图4 突变位点保守性分析及三维结构建模预测

表1 生物信息学功能预测
1.3 治疗及随访 结合患儿临床特点及基因检测结果,明确诊断为IHH 基因杂合突变致身材矮小。家属强烈要求改善患儿身高,查阅相关文献,IHH基因杂合突变无rhGH 治疗禁忌证,即予患儿rhGH 0.13 IU/(kg·d)皮下注射,维生素D 800 IU/d,并定期随诊。现已治疗3 个月,3 岁2 个月身高91.8 cm,身高增长了3 cm,治疗期间监测血常规、肝肾功能、电解质、甲状腺功能、空腹血糖胰岛素均正常,25 羟维生素D 较前上升,IGF-1 较前升高,未出现脊柱侧弯,见表2。

表2 患儿接受rhGH 治疗随诊表
2 讨论
IHH 基因位于2 号染色体q35-q36,编码IHH蛋白,在软骨板、指(趾)尖和生长板中肥大前期的软骨细胞中特异性表达。该蛋白功能涉及软骨细胞分化、关节发育与骨的形成,可在发育过程中调节骨骼的生长和骨化之间的平衡,同时可诱导甲状旁腺激素相关蛋白(parathyroid hormone-related protein,PTHrP)的表达[1]。IHH 和PTHrP 旁分泌系统在生长板功能的调节中有重要作用, 这两种旁分泌因子在生长板内形成负反馈环:IHH 增强软骨细胞的增殖和成熟,PTHrP 防止软骨细胞过早的肥大分化,IHH和PTHrP 之间的负反馈回路使软骨细胞保持增殖状态,保持软骨细胞柱的长度[3],IHH 基因突变会导致生长板的过早关闭,进而导致身材矮小。此外,IHH 基因突变导致IHH 与受体Ptch1 和拮抗蛋白Hip1 的结合减弱,下游转录因子Gli1 激活减弱,从而下调刺猬蛋白信号通路[4],导致指(趾)骨远端祖细胞的募集减少,指(趾)骨的生长受损致使指(趾)骨缩短。
除了IHH 蛋白,刺猬蛋白家族还包括另外两个序列高度保守的HH 同源物:Sonic HH(SHH)和Desert HH(DHH),它们分别对神经系统和睾丸发育至关重要[5]。IHH 基因纯合突变导致股骨头发育不良(acrocapitofemoral dysplasia,ACFD;OMIM 607778),这种疾病的特征是手部和髋部有锥形骨骺,并伴有严重不成比例的矮小[6]。IHH 基因杂合突变可导致BDA1[2],典型表现是身材矮小,中节指(趾)骨明显缩短,通常表现为中节指(趾)骨发育不全或与末端指(趾)骨融合,大拇指近侧指(趾)骨缩短,部分患儿掌骨缩短。IHH 基因杂合变异具有很大的临床异质性。Lucia 等[7]报道16 例IHH 杂合突变患儿中,均无BDA1 典型表现,其中2例患儿仅有身材矮小,9 例患儿身材矮小伴各种手指畸形(中节指骨缩短、远段指骨缩短、掌骨缩短、单独的指弯曲和锥形骨骺等),5 例患儿身材正常伴有指骨短缩。此外,第五指中节指骨短是常见的手部骨骼畸形,可单独存在。研究表明,IHH 突变人群中手部X 线观察到第五指中节指骨短发生率为64.3%,而在普通人群中的发生率为12.1%[8-9]。第五指是手部最后骨化的部位,第五指的骨骺发育不良可能与软骨形成障碍的易感性有关。本例患儿的表型要轻的多,仅表现为身材矮小伴有轻微的指骨短。
迄今报道的IHH 基因突变主要包括错义突变、移码突变等。文献表明,位于氨基末端结构域的所有突变都会影响该分子的结构,从而影响其生物学功能[10]。到目前为止,p.Glu95Lys(E58K)、p.Glu131Lys(E131K)和p.Asn100Glu(D100E)已被功能验证,这3 种变异均位于氨基末端区域,影响了Hh 与Ptc 的结合,降低其诱导细胞分化的能力[2]。本例患儿基因提示结果显示c.174G>C(p.E58D),编码区第174 号核苷酸由鸟嘌呤变异为胞嘧啶,导致第58 号氨基酸由谷氨酸变异为天冬氨酸,为错义突变。该突变位点位于氨基末端信号域,与前文相符,表明高度保守的谷氨酸对于IHH 信号通路的重要性。本例患儿突变位点为新发位点,结合临床特点及遗传学分析可判定为致病,丰富了该疾病的基因谱,蛋白功能研究对于验证临床意义未明的突变位点很重要,未来还需在此方向努力。
针对患儿身材矮小,应用rhGH 治疗是改善身高的重要方法。目前IHH 基因突变患儿应用rhGH改善身高鲜有报道。除了本研究中描述的患儿外,国内外文献中还报告了另外6 例患儿应用rhGH 改善身高(表3)[8,11]。本例患儿治疗开始年龄为2 岁10个月,为目前世界上报道应用rhGH 治疗最小年龄患儿。7 例患儿应用rhGH 治疗短期身高SDS 均有所改善。本例患儿在rhGH 治疗期间,IGF-1 水平保持在正常范围的上限,表明对rhGH 具有良好的敏感性。目前,越来越多的研究表明应用rhGH 治疗可以提高生长速度,改善矮小患儿身高。应用rhGH 治疗使IGF-1 维持在较高水平,可对生长板软骨细胞的增殖和肥大有刺激作用,在许多情况下非特异性地加速线性生长,从而部分弥补影响生长板IHH 信号通路的缺陷。但本例患儿应用rhGH 治疗时间较短,是否对终身高有改善还需长期随访。

表3 7 例应用rhGH 治疗的IHH 杂合突变患儿的基本资料
总之,本研究报道1 例以身材矮小就诊于儿科内分泌门诊的患儿,患儿为家族性矮小同时伴有手足小,指趾短粗,左手正位X 线提示第五指中节指骨短等表现。遗传学检查提示IHH 基因杂合突变。IHH 杂合突变可导致具有典型表现BDA1,还可能导致身材矮小,伴或不伴有轻微的骨骼异常,因此建议临床医师接诊家族性矮小患者时,应结合临床和影像学提示,即使症状不典型,也应及时行基因检测以进一步明确病因。此外,rhGH 可作为改善IHH 基因杂合突变患儿身高的治疗方法,但仍需大样本长时间观察其有效性和安全性。
