多联吡啶铱(Ⅲ)配合物的合成、发光及DNA/BSA结合活性*
2022-06-01顾顺心施鹏飞
顾顺心,姜 琴,施鹏飞
(江苏海洋大学 环境与化学工程学院,江苏 连云港 222005)
药物治疗是目前临床治疗肿瘤的重要手段之一,开发具有独特疗效的抗肿瘤药物一直是药物研发领域的热点。顺铂(CDDP)是最早临床应用的金属抗肿瘤药物[1],该药物因具有广谱抗肿瘤活性和高治愈率而在多种癌症治疗中表现不俗[2-4],特别是针对睾丸癌的治疗,它使睾丸癌患者的死亡率从100%降到10%以下[5]。但顺铂类药物也存在严重的毒副作用[6],且容易产生耐药性、水溶性小、在体内不易代谢,因此迫切需要开发新型金属抗肿瘤替代药物,以减少铂类药物毒副作用和交叉耐药性带来的问题。
很多铱(Ⅲ)配合物因具有较强的细胞渗透能力以及独特的抗肿瘤活性,而引起生物医药领域研究人员的广泛关注[7-9]。不仅如此,铱(Ⅲ)配合物还表现出优异的发光性能,包括激发/发射波长易于调节、量子产率高、斯托克斯位移值大、发光寿命长、光稳定性好等特点,使其在发光材料研究领域也备受青睐[10-11]。鉴于发光铱(Ⅲ)配合物所具有的光学特性,可采用激光共聚焦显微镜来追踪其在肿瘤组织内的摄入、分布与代谢状态,这将有助于研究者明确铱(Ⅲ)配合物抗肿瘤的靶分子与作用机理。另外,部分配合物在光激发下还可在肿瘤细胞内诱导生成具有强氧化性能的活性氧(ROS)物种,导致细胞坏死或凋亡。目前发光铱(Ⅲ)配合物已经作为一类非常有潜力的肿瘤光动力学治疗的光敏剂[12]而被广泛研究。
Ye等[13]的研究表明,Ir(Ⅲ)配合物对多种癌细胞表现出强抗增殖活性(强于顺铂类药物),甚至对顺铂耐药细胞株也有很好的药效;该配合物具有强磷光发射性能,利用激光共聚焦显微成像技术发现配合物能够有效地渗透到Hela细胞中;对线粒体的形态进行实时监控发现,配合物能诱导线粒体损伤从而杀死癌细胞。Yang等[14]研究表明,半三明治型Ir(Ⅲ)配合物对A549细胞表现出良好的抗癌活性(IC50=4.7 μmol/L),该配合物能靶向并破坏A549细胞的溶酶体,从而导致细胞死亡。Liu等[15]研究表明环金属化铱配合物具有双重(转移抑制和溶酶体损伤)抗癌机制:配合物通过血清白蛋白及静电力猝灭机制进行运输,且能有效地阻止癌细胞的转移,扰乱细胞周期G0/G1期,从而诱导细胞凋亡;配合物通过能量依赖性细胞摄取机制,有效积累在溶酶体中,诱导溶酶体损伤,最终导致细胞死亡。Liu等[16]将四苯乙烯基团引入Ir(Ⅲ)配位单元,得到的系列配合物对癌细胞和正常细胞具有一定的选择性;不同结构的配合物其抗肿瘤机制有所不同:部分配合物不仅可以催化NADH转变为NAD+,还可以诱导ROS富集。Lu等[17]报道了末端含芘基团的双核三联吡啶铱配合物,该配合物表现出强光毒性和相对较弱的暗毒性;配合物能在细胞内生成ROS,导致肺癌细胞A549和人皮肤癌细胞A431的线粒体和溶酶体功能损伤,以细胞凋亡的方式促使细胞坏死。Kuang等[18]报道了一种以蒽醌三联吡啶衍生物作为主配体的Ir(Ⅲ)配合物,该配合物在双光子PDT治疗厌氧肿瘤方面具有非常良好的应用前景。该配合物在缺氧条件下经还原酶还原后,发光性能显著增强,且在近红外双光子光激发下具有很高的稳定性;辐照后,配合物可以诱导产生碳自由基,促进线粒体膜电位降低与细胞死亡(光激发下IC50=2.1 μmol/L, 黑暗中IC50=58.2 μmol/L,PI=27.7);体内有A549癌细胞的雄性小鼠注射给药后,经双光子激发照射(730 nm, 50 mW, 1 kHz, 脉冲宽35 fs, 5 s/mm),2 d后小鼠体重没有明显变化,14 d后小鼠体内的肿瘤组织显著减小。Pracharova等[19]报道的配合物对顺铂耐药的肿瘤细胞表现出一定的生长抑制活性;研究发现,抗肿瘤活性并非基于DNA损伤,而是通过优先靶向内质网并抑制蛋白翻译过程从而杀死肿瘤细胞;配合物表现出良好的PDT活性,可见光照可使IC50值降低两个数量级。
1 实验部分
1.1 仪器与试剂
本文中未特别指明的化学品都来源于商业采购并直接使用,小牛胸腺DNA(ct-DNA)和牛血清蛋白(BSA)从Sigma-Aldrich公司购买。所有合成的化合物均在真空干燥器(CaSO4)中干燥后再进行结构表征。核磁数据(1H NMR)采用500 MHz Bruker DMX采集,利用四甲基硅烷(TMS)做内标。质谱数据用Agilent 1290-6545 LC/MS质谱仪采集。紫外-可见吸收光谱用UV-3100记录,荧光光谱用Hitachi F-7000记录。ct-DNA的浓度用紫外光谱标定(ε260 nm=6 600 L/(mol·cm))。在Tris-HCl的缓冲溶液中(pH=7.42),260 nm和280 nm处紫外吸光度的比值为1.87,表明DNA中已完全去除了蛋白质杂质。称取一定量牛血清蛋白BSA,用Tris-HCl缓冲液溶解,配制浓度为2×10-4mol/L的储备液,置于4 ℃保存。
1.2 合成与表征
NNC配体及其Ir(Ⅲ)配合物合成路线见图1。
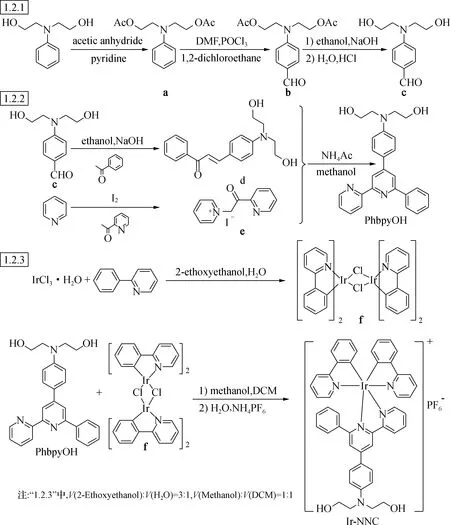
图1 NNC配体及其Ir(Ⅲ)配合物的合成路线
1.2.1N,N-二羟乙基苯甲醛的合成与表征 将52 gN,N-二(2-羟乙基)苯胺溶于70 g吡啶中,加入90 g乙酸酐后在90 ℃下搅拌24 h,冷却至室温,用400 mL水洗涤后用200 mL乙酸乙脂萃取。乙酸乙酯溶液用无水硫酸镁干燥,减压蒸除溶剂,得油状物N,N-二乙酸乙酯基苯胺(化合物a)。1H NMR(CDCl3,500 MHz),δ:7.2(2H),6.8(3H),4.3(4H),3.6(4H),2.1(6H)。
将40 g POCl3在0 ℃搅拌滴入50 mL DMF中,溶液变为淡红色油状物。0 ℃继续搅拌15 min后,将50 gN,N-二乙酸乙酯基苯胺溶于30 mL 1,2-二氯乙烷并滴入淡红色油状物中,滴完后升温至90 ℃,搅拌3 h得墨绿色混合溶液。冷至室温后,倒入碎冰中搅拌,用质量分数30%的K2CO3溶液将溶液pH调节至6~8,得到黄色固体N,N-二乙酸乙酯基苯甲醛(化合物b)。1H NMR(CDCl3,500 MHz),δ:9.6(1H),7.7(2H),6.8(2H),4.2(4H),3.6(4H),2.0(6H)。
取15 gN,N-二乙酸乙酯基苯甲醛溶解于含有12.5 g氢氧化钠的250 mL乙醇水溶液(乙醇与水体积比为175∶75)中,搅拌7 h后用稀盐酸(4 mol/L)调节pH至8~9。将溶液蒸干后用乙酸乙酯洗涤3遍,蒸除乙酸乙酯,得黄色油状固体。柱层析(硅胶柱,展开剂为体积比1∶1的乙酸乙酯和石油醚)后得到黄色粉末N,N-二羟乙基苯甲醛(化合物c),产率90%。1H NMR(CDCl3,500 MHz),δ:9.65(1H),7.71(2H),6.71(2H),3.91(5H)。
1.2.2 配体PhbpyOH的合成与表征 将2.9 g(14 mmol)N,N-二羟乙基苯甲醛、1.68 g(14 mmol)苯乙酮和2.24 g(56 mmol)质量分数10%的NaOH水溶液在常温下搅拌12 h后,用二氯甲烷萃取,无水硫酸镁干燥后,旋蒸去除溶剂,得到2.2 g红色油状物(化合物d),产率为50.57%。
将11.8 g(50 mmol)I2溶于72 mL吡啶中,加热到60 ℃至完全溶解,再加入7.1 g(58.7 mmol)2-乙酰基吡啶,升温至95 ℃,反应3 h,静置冷却后析出固体。将固体滤出后用CH2Cl2洗涤,得到7.28 g灰色固体吡啶嗡盐(化合物e),产率44.7%。
将2.2 g(7 mmol)化合物d和2.3 g(7 mmol)化合物e溶于100 mL甲醇,回流反应30 min后分3批加入3.27 g NH4Ac,继续回流24 h后静置冷却,用乙酸乙酯萃取后干燥,旋蒸除去溶剂得棕色油状物。柱层析(硅胶柱,展开剂为体积比4∶1的乙酸乙酯和石油醚)后得到黄色粉末(化合物PhbpyOH),产率为14%。1H NMR(CDCl3,500 MHz),δ:8.70(t,1H),8.67(d,1H),8.57(d,1H),8.20(d,2H),7.93(d,1H),7.88(td,1H),7.74(d,2H),7.47(t,1H),7.38~7.31(m,1H),6.77(d,2H),4.17~3.96(m,2H),3.88(t,4H),3.64(t,4H)。
1.2.3 配合物Ir-NNC的合成与表征 将397 mg(2.56 mmol)的2-苯基吡啶、764 mg(1.28 mmol)的三氯化铱水合物和20 mL乙二醇水溶液(V(乙二醇)∶V(水)=3∶1)的混合溶液置于Schlenk管中,N2保护下120 ℃加热回流24 h。反应结束后冷却至室温析出沉淀,离心过滤得到黄色粉末,依次用水(3×20 mL)、乙醇(3×10 mL)和乙醚(3×10 mL)分别洗涤,真空干燥后得到543.8 mg[Ir(bpy)2Cl]2(化合物f)。
将100 mg(0.093 mmol)化合物f和76.57 mg(0.186 mmol)PhbpyOH溶于30 mL二氯甲烷与甲醇的混合溶液(V(二氯甲烷)∶V(甲醇)=1∶1)中,65 ℃下回流反应24 h,全程N2保护且避光处理。反应结束后减压蒸除溶剂,得到橙色固体。将橙色固体溶于30 mL水中,加入150 mg NH4PF6固体后继续搅拌30 min,过滤收集橙色沉淀物,依次用水(3×20 mL)、乙醇(3×10 mL)和乙醚(10 mL)洗涤,真空干燥后得到橙色粉末(化合物Ir-NNC)72.4 mg,产率为37.2%。表征结果为1H NMR(d6-丙酮,500 MHz),δ:9.07(d,1H),9.03(d,1H),8.28(td,J=8.1 Hz,1H),8.11(dd,J=8.5 Hz,2H),8.01(ddd,5H),7.93(td,J=7.9 Hz,1H),7.76(d,1H),7.70(dd,1H),7.65~7.59(m,1H),7.41~7.36(m,1H),7.27(ddd,1H),7.23~7.17(m,1H),7.01~6.94(m,3H),6.92(td,1H),6.86~6.60(m,5H),6.58~6.53(m,1H),6.35(td,1H),6.02(dd,1H),5.65(dd,1H),4.23(t,2H),3.89~3.73(m,4H),3.70(t,4H)。ESI-MS(+p),m/z=912.289 1,可指认为[Ir(ppy)2PhbpyOH]+的分子离子峰。
1.3 单晶结构分析
配体PhbpyOH的单晶通过其在溶剂(V(乙酸乙酯)∶V(石油醚)=1∶1)中缓慢挥发得到。选取表面光滑、无裂纹、大小合适的单晶,置于带有石墨单色器的Bruker APEX CCD X-ray衍射仪上,于296 K下使用MoKα(k=0.071 073 nm)射线收集衍射数据,使用Bruker APEX2[20]和Bruker SAINT[21]程序进行衍射数据的收集和还原。使用Olex2[22]软件进行晶体解析。Olex2.Solve程序对配体PhbpyOH晶体结构进行初解, 并使用SHELXTL-2018[23]程序对晶体结构进行精修获得非氢原子坐标和各向异性参数,氢原子由理论计算方法得到。CCDC:2125620。
1.4 DFT理论计算
配体和配合物的DFT计算采用Gaussian 09程序包进行,利用B3LYP密度泛函理论对分子结构进行了优化。在基态和激发态优化的基础上,运用TDDFT方法研究了激发态的电子性质。在计算过程中,配体的所有原子采用 6-31g(d)基组,配合物采用了6-31g(d)(H,O,N,C)/LANL2DZ(Ir)混合基组。
1.5 单线态氧量子产率
在体外使用9,10-蒽二基-二(亚甲基)二丙二酸ABDA作为1O2指示剂,用功率为6 W的紫外灯在365 nm处激发配合物Ir-NNC(初始浓度为10 μmol/L)的乙腈溶液,每间隔10 s记录一次ABDA的吸光度(418 nm处)。采用不含配合物的ABDA的乙腈溶液作为对照试验,采用Ru(bpy)3Cl2(ΦΔ=0.56)敏化ABDA为参考,计算Ir-NNC敏化ABDA的量子产率。量子产率计算公式为
其中:sam表示配合物Ir-NNC;std表示Ru(bpy)3Cl2;S表示ABDA吸光度图的斜率;F表示吸收校正因子,F=1-10-OD(OD为Ru(bpy)3Cl2和配合物的光密度)。
1.6 双光子荧光光谱
双光子激发荧光光谱使用锁模钛宝石激光器(Coherent Mira900F)作为激发光源,脉冲持续时间200 fs,重复频率76 MHz,采用单扫描条纹相机(Hamamastu C5680-01)及单色仪记录双光子荧光信号。样品溶于DMSO,浓度1.0×10-3mol/L。
1.7 与DNA和BSA相互作用的分子模拟
采用Autodock程序对配合物与DNA(或BSA)之间的相互结合进行模拟。Ir-NNC的结构通过DFT(B3LYP)方法优化得到,DNA(PDB ID 1BNA)和BSA(PDB ID 4f5sBSA)的分子结构从蛋白质数据库中获得(http://www.rcsb.org/pdb)。在建模时,DNA-Ir-NNC的网格盒大小为6.8 nm×7.6 nm×12.6 nm,网格间距为0.037 5 nm。BSA-Ir-NNC的网格盒大小为15.0 nm×6.6 nm×10.4 nm,网格间距为0.1 nm。运用拉马克遗传算法(LGA)计算结合在DNA/BSA上的Ir-NNC可能出现的构象,进行100次GA运算,其他参数使用默认值。利用Pymol软件分析结合自由能最低的构象的对接结果。
2 结果与讨论
2.1 配体与配合物的结构
配体分子的X-ray单晶衍射结构如图2a所示,配合物经DFT优化的结构如图2b所示。
从图2a可以看出,纯配体PhbpyOH中的两个吡啶环与苯环具有一定的共平面性,最大偏差为24.63°。而在配合物Ir-NNC中,由于配位中心的位阻效应,PhbpyOH中的苯环平面与联吡啶平面的夹角增大,达到74.75°。从Ir-NNC的DFT优化的结构(图2b)可以看出,PhbpyOH中的苯环没有参与配位,这与质谱结果是一致的。Ir(Ⅲ)配位中心采取畸变的正八面体结构,其中的Ir—N键长与文献[24]报道的结果接近。

a 配体的单晶衍射结构
2.2 配体与配合物的吸收光谱
配体和配合物的紫外-可见吸收光谱见图3。配体PhbpyOH在250~400 nm范围内呈现出3个吸收带,分别是不同共轭体系之间的π→π*跃迁所致。配合物中n→π跃迁的吸收带的强度明显增加且发生了一定的红移。与Ir(Ⅲ)配位后,配体PhbpyOH中的π→π*跃迁强度显著降低,且配合物在415 nm处新出现一个强吸收峰,可归属为金属到配体的荷移跃迁(MLCT)。

图3 配体和配合物的紫外-可见吸收光谱
为了进一步明确配体和配合物中激发态的能级与结构,采用TDDFT进行了结构优化和分子轨道能量计算,结果如图4所示。PhbpyOH的HOMO轨道电子云主要集中在N,N-(2羟乙基)氨基、苯基、多联吡啶基团的中心吡啶环区域内,LUMO轨道电子云主要集中在联吡啶部位,分子中的N,N-(2羟乙基)氨基是强给电子基团,而联吡啶基团起到吸电子的作用,这种D-π-A结构有利于分子内的电荷转移。结合上述紫外-可见吸收光谱图可知,261 nm处的吸收带可归属于多吡啶基团的π→π*的跃迁吸收(HOMO-1→LUMO),而345 nm处的吸收带则归属于配体整个分子的π→π*跃迁吸收(HOMO→LUMO)。在配合物Ir-NNC中,HOMO轨道电子云主要集中在PhbpyOH的联吡啶区域上,HOMO-2轨道电子云主要集中在PhbpyOH的N,N-(2羟乙基)氨基、苯基基团上,而LUMO轨道电子云主要集中在苯基吡啶Ir(Ⅲ)配位单元上,HOMO→LUMO表现出明显的MLCT特性。
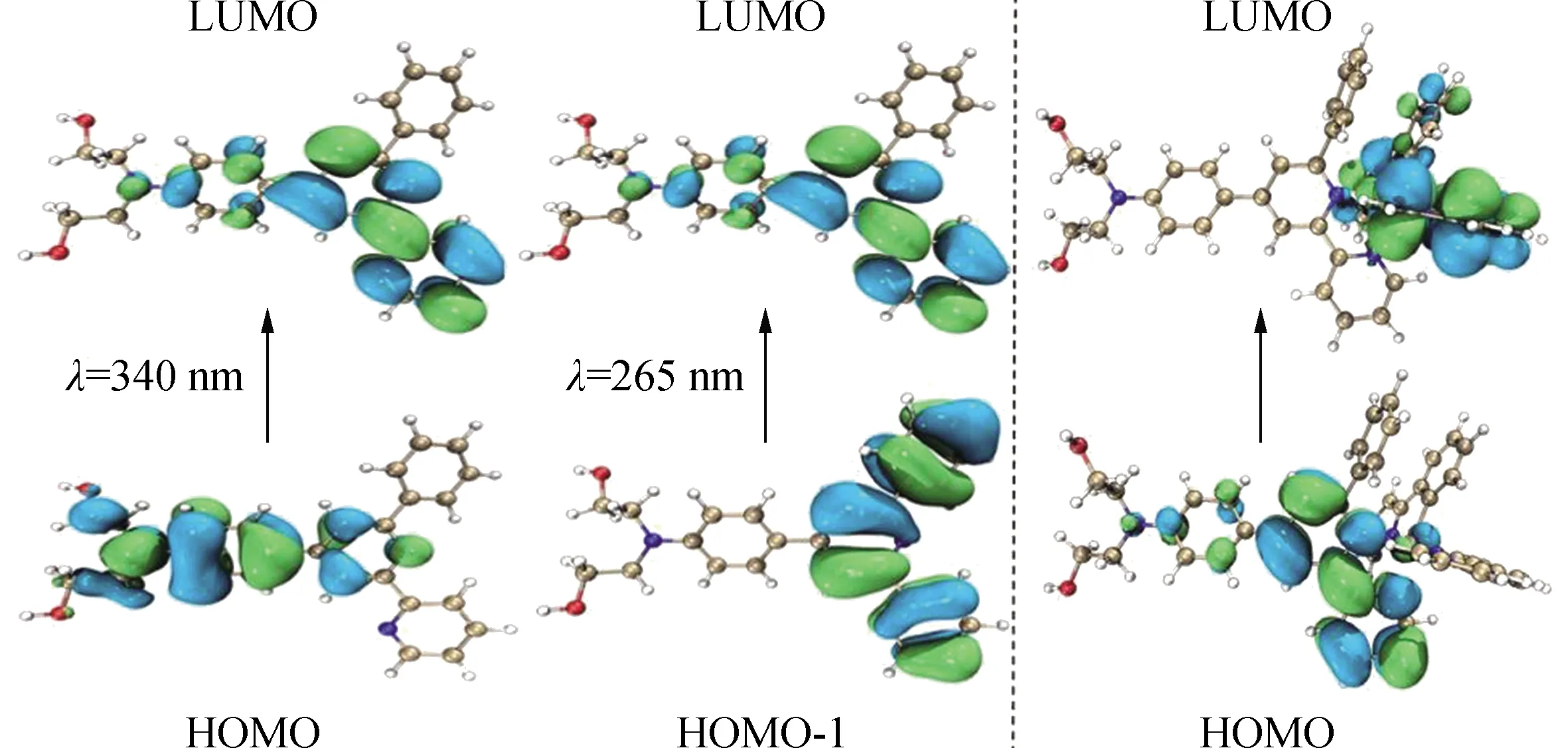
图4 配体和铱配合物的分子轨道
2.3 配体与配合物的单、双光子发射光谱
配体PhbpyOH和配合物Ir-NNC在多种溶剂中都呈现出较强的荧光发射,如图5所示。对比化合物在不同极性的溶剂中的荧光发射峰,发现它们都呈现明显的溶致变色现象,表明配体和配合物的激发态都具有一定的极性且都呈现出电荷转移发光。溶剂极性的增加促使溶剂分子与溶质分子的偶极—偶极相互作用增强,使激发态的能量降低,造成荧光光谱发生红移。与配体PhbpyOH(λem=518 nm,DMSO)比较,在同一溶剂中配合物Ir-NNC(λem=621 nm,DMSO)的最大荧光发射波长发生了明显的红移,而且荧光发射强度降低约1/2。表明Ir(Ⅲ)的配位对配体的电子云结构产生了巨大的影响。
a 配体
不仅如此,配体和配合物在760 nm的飞秒激光激发下,还表现出双光子激发荧光效应(TPEF),如图6所示。相同的测试条件下,配合物Ir-NNC在DMSO溶液中的双光子激发荧光相较配体PhbpyOH的峰值红移约40 nm,但发光强度要明显弱于PhbpyOH。配体PhbpyOH在DMSO中的最大TPEF发射波长位于528 nm,与其在DMSO中的单光子激发荧光(OPEF)相比,红移了约10 nm,这主要是由于在TPEF测量时样品浓度远大于OPEF的(约为150倍),造成了荧光的再吸收作用所致[25]。配合物Ir-NNC的TPEF峰位于567 nm处,其相对于OPEF谱带的峰蓝移了50 nm,说明单光子激发与双光子激发具有不同的激发态能级和结构。TPEF来源于能级更高的单线态激发态1MLCT,而OPEF则源自低能级的三线态激发态3MLCT。

a 配体
2.4 与DNA和BSA的相互作用
研究发现,大多数铱配合物的抗肿瘤作用都是通过光诱导产生具有细胞毒性的1O2,从而杀死肿瘤细胞[17]。使用ABDA作为1O2指示剂并以Ru(bpy)3Cl2为参照,测试了配合物Ir-NNC的相对1O2量子产率。随着光照时间的增加,含有Ir-NNC的ABDA溶液的吸光度呈现出一定的下降趋势,这表明在体外铱配合物Ir-NNC可在光照下诱导生成1O2,其量子产率约为0.1。
临床使用的许多抗癌药物都以DNA为作用靶点,它们与DNA的相互结合可引起DNA复制障碍,从而抑制癌细胞的分裂[26]。研究铱配合物与DNA分子的相互作用,对于明确其抗肿瘤的生物靶分子具有重要意义。由图7a可以看出,随着ct-DNA浓度逐渐增加,Ir-NNC的紫外-可见吸收光谱表现出明显的减色效应,但并无明显的红移效应,说明配合物不是通过嵌入作用与DNA结合[27]。而当配合物结合到DNA的沟区时,仅出现减色效应,可能是配合物的发色团被DNA的碱基覆盖所导致[28]。
血清白蛋白是血浆中含量最为丰富的载体蛋白,能与进入血液中的大多数内源性和外源性化合物结合,从而在体内起到存储和转运的作用。研究铱配合物与血清蛋白之间的相互作用[29],有助于了解配合物在体内的运输和分布情况,对于阐明铱配合物抗肿瘤的作用机制具有重要意义。由图7b可知,随着BSA浓度逐渐增加,275 nm处的吸收峰出现增色效应,这是BSA分子中色氨酸和酪氨酸的特征吸收峰所致。在300 nm和425 nm处配合物的紫外-可见光谱吸收峰均表现出明显的减色和红移现象,并在297 nm出现等吸收点,表明配合物Ir-NNC可结合到BSA上并形成Ir-NNC-BSA复合物。
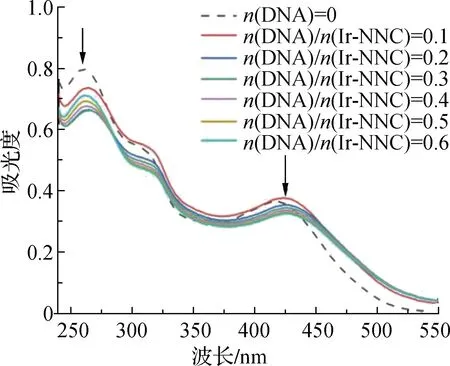
a DNA
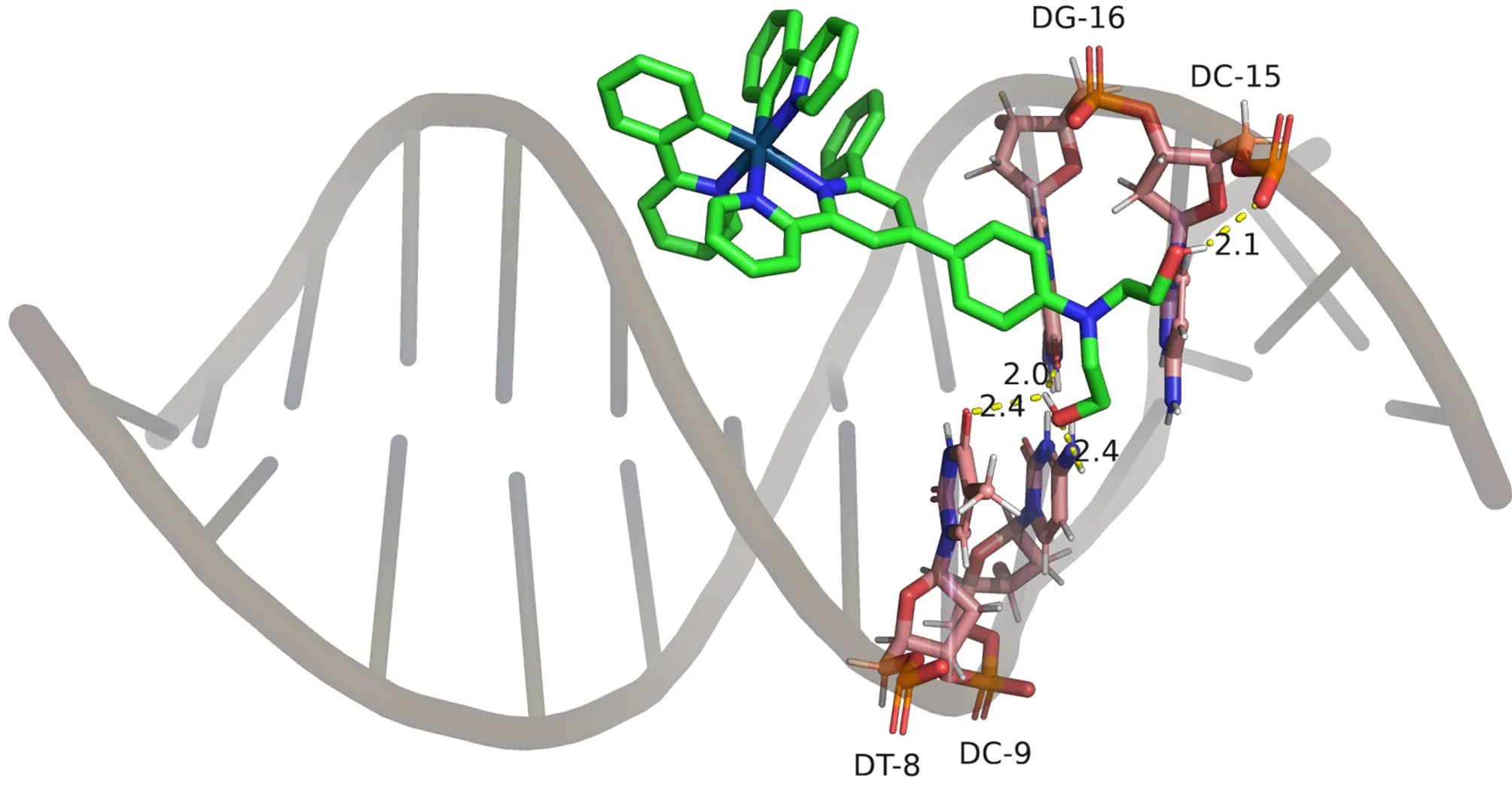
Dock分子建模法是用于探索小分子与生物大分子的结合位点和结合能的一项重要技术[30-31]。为了进一步明确Ir-NNC与DNA和BSA的结合位点,本文利用Autodock模拟了配合物与DNA和BSA之间的相互作用。对接结果表明,铱(Ⅲ)配合物Ir-NNC进入DNA的小沟区而不是插入DNA碱基对之间(这与紫外滴定的结果一致),结合自由能为-20.75 J/mol。如图8a所示,Ir-NNC的2个羟乙基与DNA行成4个氢键,分别是H92与DNA的DC-15(0.21 nm),H97与DT-8(0.24 nm),DG-16(0.20 nm),O56与DC-9(0.24 nm)。由图8b可以看出,Ir-NNC在BSA中的结合位置为ⅠB亚结构域,该结合自由能为-14.98 J/mol。图8c可知,Ir-NNC的H97和H92分别与氨基酸残基ASP-118(0.25 nm)和GLU-125(0.19 nm,0.27 nm)形成了3个氢键。氢键的存在可以稳定复合物Ir-NNC-BSA的结构。
a 与双螺旋DNA的对接(单位:Å)
