云南拉市海高原湿地H9N2 亚型禽流感病毒的遗传特性分析
2022-05-26张振兴薛晓岩王雯慧韩联宪代红炀李高银段博芳张文东李素华宋建领
季 佳,张振兴,薛晓岩,王雯慧,韩联宪,代红炀,李高银,段博芳,张文东,刘 强,李素华*,宋建领*
(1.西南林业大学生命科学学院/国家高原湿地研究中心,云南 昆明 650224;2.云南省畜牧兽医科学院云南省热带亚热带动物病毒病重点实验室,云南 昆明 650224;3.会泽黑颈鹤国家级自然保护区管护局,云南 曲靖 654200;4.昭阳区动物卫生监督所,云南 昭通 657000;5.云南省动物疾病预防控制中心,云南 昆明 650034)
禽流感病毒(Avian influenza virus,AIV)属于正黏病毒科流感病毒属的A 型流感病毒,其基因组由8 个节段的单股负链RNA 组成,可编码11 种不同的蛋白[1],其中表面蛋白血凝素(Haemagglutinin,HA)和神经氨酸酶(Neuraminidase,NA)的组合可以产生不同的亚型。自1998 年以来,H9N2 亚型AIV 已在我国大部分地区持续流行20 多年,病毒在流行过程中持续进化,基因遗传特征产生一定的变异,形成具备感染哺乳动物的能力或导致致病力增强,甚至成为H5N1(如A/Hong Kong/156/97 株)[2]、H7N9(如A/Shanghai/1/2013)[3]、H10N8(如A/Jiangxi-Donghu/346/2013 株)[4]等可感染人的流感病毒的内部基因供体。此外,当同一细胞感染两个或者多个不同的病毒时,H9N2 AIV 各亚群或与其他亚型AIV 的8 个基因节段之间可进行广泛的基因重组,产生一系列基因型特异的新亚型病毒,目前我国国内流行的H9N2 AIV 已进化出A~W 等众多的基因型[5]。
野生水禽尤其是雁形目、鸻形目的野禽被认为是AIV 的天然宿主,病毒在野禽肠道上皮细胞高效复制,通过粪便污染水源进行AIV 的有效传播[6]。云南拉市海高原湿地自然保护区于2004 年被列入“国际重要湿地”,不仅是候鸟迁徙通路上的重要停歇地,也是众多候鸟的重要越冬地,水禽资源丰富,目前已记录到的17 目44 科236 种鸟类中,水禽有89 种,约10 万只[7]。在云南拉市海高原湿地开展野禽及家禽的AIV 监测工作极其重要,本实验在AIV 监测研究中,分离到3 株H9N2 AIV,通过生物信息学分析探究了分离株的基因遗传进化及分子变异特征,为揭示AIV 在野禽和家禽中传播及掌握病毒分子进化趋势提供基础数据。
1 材料与方法
1.1 主要试剂 病毒RNA 提取试剂盒(QIAamp®Viral RNA Mini Kit)购自德国QIAGEN公司;一步法RT-PCR试剂盒(PrimeScriptTMOne Step RT-PCR Kit)购自宝生物工程(大连)有限公司;青霉素和链霉素购自上海碧云天生物技术有限公司;庆大霉素和制霉菌素购自上海阿拉丁生化科技股份有限公司;样品保存液为pH7.0~pH7.4,含青霉素2 000 IU/mL、链霉素2 mg/mL、庆大霉素50 μg/mL 及制霉菌素1 000 IU/mL 的PBS 溶液;SPF鸡胚购自北京妫川亚申养殖中心有限公司。
1.2 样品采集 于2020 年3 月~2021 年3 月在云南拉市海高原湿地采集到实验样品1 807 份,其中野禽新鲜粪便样品1 367 份,包括:红嘴鸥500 份、斑头雁450 份、灰雁387 份和灰鹤30 份,家禽泄殖腔拭子样品420 份以及环境水样20 份。野禽粪便样品采集时,通过高倍望远镜远距离观察拉市海高原湿地滩涂的野禽种类及大致种群数量,重点跟踪斑头雁、灰鹤、灰雁、黑颈鹤及红嘴鸥等涉水野禽的种群。选择1 h内的新鲜粪便,根据粪便形态特征确定来源的野禽种属后,立即置入盛有样品保存液的冻存管。家禽泄殖腔样品来自拉市海湿地保护区缓冲区外的家禽散养户养殖的无临床症状的鸡。环境水样包括野禽生境地的沼泽水以及散养户的养殖排放污水。盛有样品的冻存管,放入干冰中,24 h 内运回实验室。
1.3 病毒分离 采集的粪便样品、泄殖腔拭子和水样经研磨及涡旋振荡后,8 000 r/min 离心3 min,取上清液加入青霉素(工作浓度2 000 IU/mL)和链霉素(工作浓度2 mg/mL),放置4 ℃冰箱过夜处理。将处理后样品按每胚200 μL 经尿囊腔接种于10 日龄SPF鸡胚,每份样品接种5 枚,置37 ℃恒温箱孵育,收取死胚及96 h 活胚的尿囊液,盲传2 代后,测定其血凝活性,无菌收集血凝效价高于4 log2 的鸡胚尿囊液,视为AIV 可能阳性的样品。
1.4 分离病毒的RT-PCR 扩增 提取血凝阳性鸡胚尿囊液的病毒RNA,参考文献[8]合成AIV M基因检测引物AIF/AIR,参考文献[9]合成可同时扩增AIV全基因组8个节段的引物MBTuni-12/13,经一步法RT-PCR分别扩增M基因和8个基因节段。M基因扩增的反应程序为:50 ℃30 min;94 ℃2 min;94 ℃30 s、45 ℃30 s、72 ℃1 min,35个循环;72 ℃10 min。AIV 8个基因节段扩增的反应程序为:42 ℃60 min;94 ℃2 min;94 ℃30 s、45 ℃30 s、68 ℃3 min,5 个循环;94 ℃30 s、57 ℃30 s、68 ℃3 min,31个循环。以本实验室保存的A/chicken/Yunnan/1/2019(H9N2)(简称CK/YN/1/19)株作为阳性对照,以不加模板的反应体系作为阴性对照。
1.5 分离病毒RT-PCR 产物的测序分析 RT-PCR产物经回收纯化后由上海伯豪生物技术有限公司进行二代测序分析。扩增产物利用Illumina 公司nex⁃tRAD 法构建测序文库,通过Illumina Miseq 测序平台完成测序,采用CLC Genomics Workbench 对测序读段Reads 组装的Contigs 进行拼接,从而得到编码蛋白基因(Unigens)的序列集合[10],将基因核酸序列翻译为氨基酸序列,在GenBank 的非冗余蛋白质数据库(Non-redundant protein databasse,NR)进行同源性比对,获取AIV 蛋白的相似性序列。
1.6 分离病毒的生物信息学分析 成功注释为AIV各基因节段的序列通过NCBI BlAST 工具进行同源性比对,分析与8 个节段相似性较高的AIV,并下载H9N2 AIV 参考序列,以MEGA 7.0 的Clusta W 多序列比对和邻近法(Neighbor-joining,NJ)构建遗传进化树,可靠性通过1 000 Bootstrap 值评估,利用Laser⁃gene 软件将分离株8 个基因节段翻译为蛋白质序列,分析流感病毒关键氨基酸位点突变情况,并通过NetNGlyc 1.0 server预测HA和NA蛋白糖基化位点。
2 结 果
2.1 病毒的分离鉴定结果 将经血凝试验鉴定为AIV阳性样品M 基因的RT-PCR 扩增结果显示,有3 份样品出现预期目的条带(图1A),初步表明其为AIV阳性;其中2 份样品为家禽泄殖腔拭子,1 份为斑头雁粪便样品,家禽样品的AIV 阳性分离率为0.48%(2/420),野禽样品的AIV 阳性分离率为0.07%(1/1367)。3 株AIV M 基因阳性分离株均扩增到AIV 全基因组的8 个节段(图1B),8 个基因节段经测序及NCBI BLAST 分析结果表明,3 株分离株均为H9N2 亚型AIV,分别命名为:A/chicken/Yunnan/LSH14/2021(H9N2)(简称CK/YN/LSH14/21 株)、A/chicken/Yunnan/LSH16/2021(H9N2)(简称CK/YN/LSH16/21 株)、A/bar headed goose/Yunnan/LSH142/2021(H9N2)(简 称BHG/YN/LSH142/21株)。
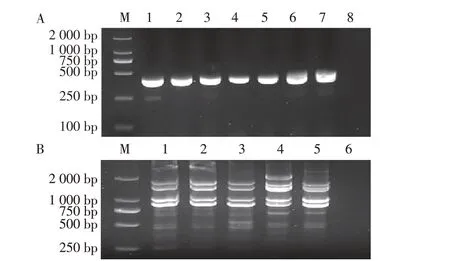
图1 RT-PCR扩增AIVs M基因和全基因组Fig.1 RT-PCR amplification of M gene and whole genome of AIVs
2.2 分离病毒8 个基因节段的遗传进化分析 核苷酸同源性分析结果显示,3 株H9N2 AIV 分离株HA基因的同源性为97.1%~97.4%,与A/chicken/Guangxi/55/2005(H9N2)同源性为92.3%~92.6%;NA 基因的同源性为97.5%~98.6%,与A/chicken/Jiangsu/WJ57/2012(H9N2)疫苗株同源性为91.3%~91.4%。将分离株与H9N2 AIV 代表株进行遗传进化分析,结果显示,3株分离株的HA基因均位于h9.4.2.5分支(图2),NA 基因均位于n2.1.4分支(图3),这两个分支均属于欧亚谱系的Y280-like 亚群,PB2、M 基因属于欧亚谱系的G1-like亚群,PB1、PA、NP和NS基因属于欧亚谱系的F/98-like亚群,由此可推测3株分离株均为重组病毒,且与我国流行的H9N2 AIV S基因型重组模式一致(图4),与早年流行的A 和H 基因型相比,S 基因型在家禽中具有更强的传染性[5]。研究显示具有G1-like 特性的M 基因可增加AIV 对人的感染[11],CK/YN/LSH14/21分离株M 基因核苷酸序列与人源A/Beijing/1/2017(H9N2)株M基因序列同源性高达99.30%,提示禽源H9N2 AIV可作为重组人源病毒的供体。
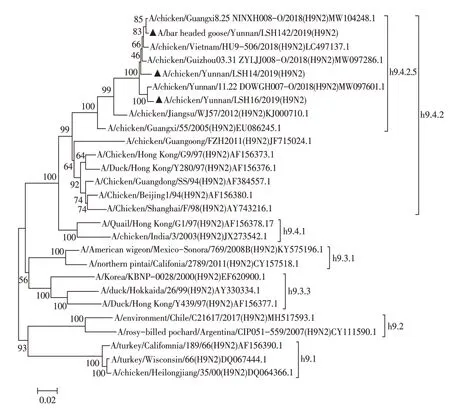
图2 3株H9N2分离株HA基因的遗传进化树Fig.2 The phylogenetic tree of the HA genes of the three H9N2 isolates
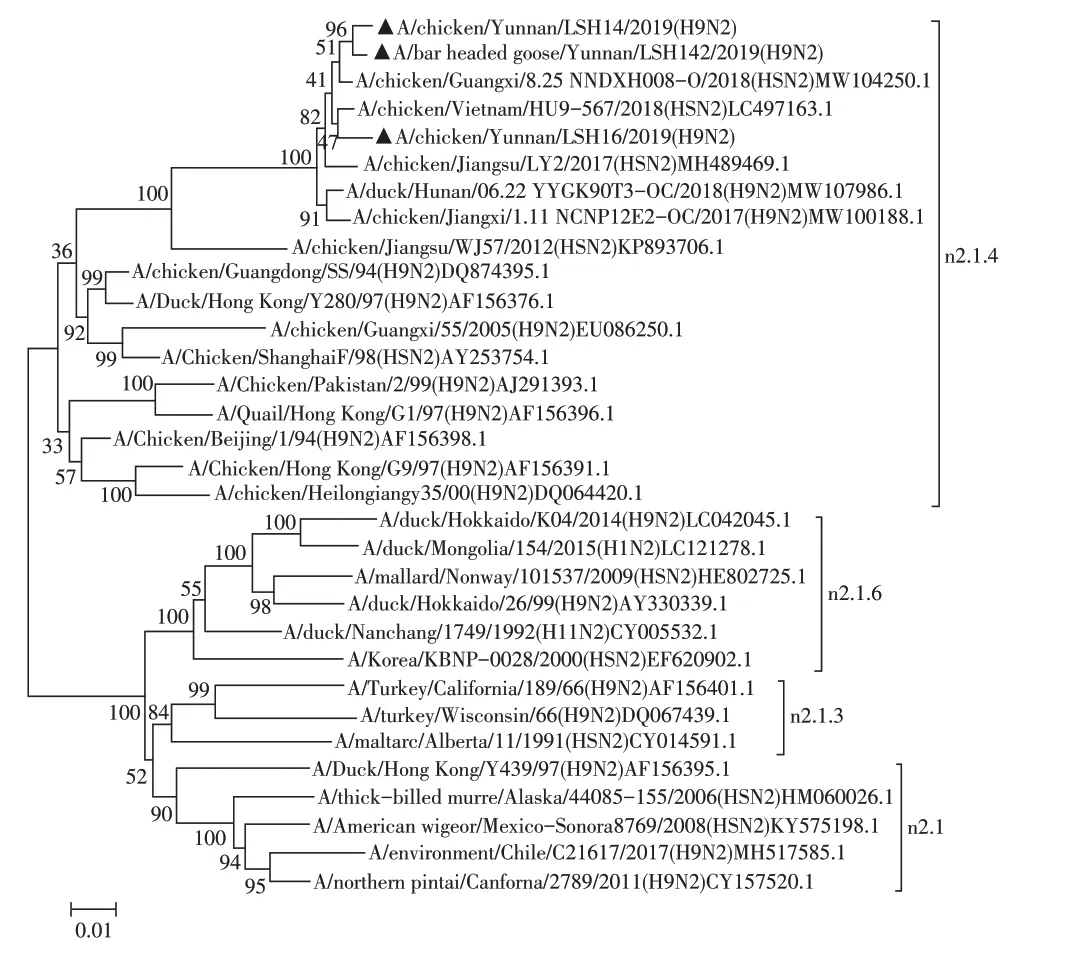
图3 3株H9N2 AIV分离株NA基因的遗传进化树Fig.3 The phylogenetic tree of the NA genes of the three H9N2 isolates

图4 3株H9N2 AIV分离株的S基因型重组模式Fig.4 The S genotype recombination model of the three isolated viruses
2.3 分离病毒8 个基因节段的特征性氨基酸位点的分析 将3 株H9N2 AIV 分离株与参考株A/Chicken/Beijing/1/94(H9N2)、A/Chicken/Shanghai/F/98(H9N2)和A/chicken/Guangxi/55/2005(H9N2)比对,进行流感病毒特征性氨基酸位点分析(表1)。HA 蛋白氨基酸特征性位点分析结果显示,3 株H9N2 AIV HA 蛋白的裂解位点均为333PSRSSR↓GLF341,无连续的碱性氨基酸(K、R、H),表明3 株H9N2 AIV 符合低致病性AIV 的 特 征[12];HA 蛋 白 的Y109、W161、T163、N191、L202、Y203(H9 编号)等氨基酸位点高度保守,但发生了疫苗接种地区流行株特征性N166D 突变以及增加跨种感染哺乳动物能力的A198T 和Q234L 变异[13-14],其中A198T 则可增强病毒与受体的亲和力,Q234L 变异则使得病毒具有与人上呼吸道上皮细胞表面的唾液酸α-2, 6 半乳糖(SA α-2, 6 Gal)受体结合的特性;由于HA 蛋白发生T220I、P315S 突变,导致218 位糖基化位点缺失,在313 位点新增一个糖基化位点,两个糖基化位点的改变可微弱增强AIV 在组织中的复制能力[15]。

表1 3株H9N2 AIV分离株的病毒传播能力及与致病力相关氨基酸位点分析Table 1 Analysis of the special amino acid sites associating with viral pathogenicity and transmission of the three isolated viruses
NA 蛋白氨基酸特征性位点分析显示,颈部aa63~aa65 位点缺失,这种缺失是中国大陆鸡源H9N2 AIV 的标志[16],提示本实验中分离到的野禽来源的分离株可能来自家鸡感染,这种氨基酸位点的缺失也导致NA 蛋白产生Y280-like 亚群特征性的61位糖基化位点缺失[17]。
3 株分离株内部基因编码蛋白的特征性氨基酸位点分析显示,PB2 蛋白存在与小鼠致病力增强相关的L89V、G309D、T339K 氨基酸替换[18],但未发现与跨越物种屏障感染哺乳动物以及致病力增强相关的E627K 和D701N 突变;此外3 株H9N2 AIV 分离株的PB1蛋白D622G、M1 蛋白N30D 和T215A、M2 蛋白S31N 以及NS1 蛋白P42S 等与病毒致病力增强的位点均发生了突变,这些内部基因的突变在H9N2 AIV 的适应性进化中至关重要[19-21]。本实验中分离到的野禽源H9N2 AIV 与2 株家禽源AIV 的分子特征仅在PA 蛋白N383D 的突变具有差异性,这种突变可增强病毒聚合酶活性的突变[22],野禽源H9N2 AIV 无该突变。
3 讨 论
世界范围分布的H9N2 AIV,在遗传进化上可依据地域分布特征分为北美谱系和欧亚谱系两大分支[2]。2012 年我国研究者对GenBank 数据库中千余条H9 亚型AIV 的HA 序列分析,将其分为h9.1~h9.4 4 个进化分支[23]。h9.1 和h9.2 分别为上世纪60 年代和90 年代的北美地区分离株;h9.3 分支为上世纪70年代持续至今的分离株,覆盖的地域包括亚洲、欧洲、非洲、太平洋和北美等地;h9.4 分支则对应从1994 年以来亚洲流行的分离株,也是最为庞大的分支,包括两个亚分支h9.4.1(即以A/quail/Hong Kong/G1/97 株为代表的G1-like 亚群)和h9.4.2(即以A/duck/Hong Kong/Y280/97 株 为 代 表 的Y280-like 亚 群)。h9.4.2 全部是中国的分离株,可进一步进行四级细分,其中h9.4.2.1~h9.4.2.4 进化分支代表2007 年之前国内H9N2 AIV 分离株,2007 年出现了我国日趋流行以A/chicken/Guangxi/55/2005 株为代表的h9.4.2.5 分支,2010 年又出现了以A/chicken/Guangdong/FZH/2011 株 为 代 表 并 呈 蔓 延 趋 势 的h9.4.2.6 分 支,但 当前我国仍以h9.4.2.5 分支的流行株为主。基于AIV 各基因系统发育的基础,我国研究者将H9N2 AIV 在中国的演变聚类成具有明显时空特征的A~W 基因型,其中在中国鸡群中A、H 和S 型H9N2 AIV 分别在不同时期占据主导地位[5]。研究者将A 基因型定义为分离株全部基因均来自BJ/94-like;H 基因型定义为分离株的3 个聚合酶基因PA、PB1、PB2 以及NP 基因均来自F/98-like 而其他基因来自BJ/94-like;S 基因型是在H 型基础上出现的由G1-like 提供PB2 和M 基因;90 年代早期,我国以A 基因型流行为主,2000 年开始流行在家禽中具有更好适应性的H 基因型,2007 年以后S 基因型逐渐在鸡群中稳定流行。流行病学研究表明我国当前流行的H9N2 AIV 仍为S 基因型[24]。
本实验分离自云南拉市海高原湿地的野禽及家禽来源的H9N2 AIV 属于我国当前流行的h9.4.2.5 分支,同属于S 基因型。值得注意的是这株分离自斑头雁的H9N2 AIV 也具有当前中国鸡源AIV 标志性的NA 蛋白茎部结构中aa63~aa65 的缺失[16],同时存在疫苗接种地区流行株特征性HA 蛋白N166D 突变[13],推测本实验分离的野禽源AIV 可能来自家禽的传播感染。本研究揭示AIV 可能存在由家禽向野禽传播,为家禽养殖业的生物安全防护提出了更高的要求。
野禽导致的禽流感疫情风险受到了研究者的普遍关注。野生水禽和家养水禽通过水环境密切接触,易将AIV 传播给家禽,2015 年发生在欧洲的H5N8 高致病性AIV 经证实即是由野生水禽传染给家禽[25],尤其是迁徙性涉水禽类通过长距离的迁徙活动在全球AIV 传播中起着重要作用[26]。云南拉市海高原湿地位于中亚迁徙线和东亚-澳大利亚迁徙线两条全球候鸟通道的交汇处,由于独特的地理位置和适宜的生境,每年冬季大量迁徙野鸟在此越冬或停歇,野鸟种群数量约10 万只[7]。斑头雁、灰雁、黑颈鹤和灰鹤等涉水野禽均有较大的种群数量在此越冬,斑头雁种群数量约7 万只,在全球分布广泛,每年可沿着中亚迁徙线飞越喜马拉雅山脉,在中亚和南亚之间长距离迁徙[27-28]。处于候鸟迁徙路线的重要十字路口,拉市海高原湿地丰富的野生水禽资源,带给家禽的流行病学风险以及对人类健康的潜在威胁不容忽视。
本实验中的3 株H9N2 AIV 分离株与我国其它地区分离到同型病毒存在相似的分子变异趋势,如与人源H9N2 AIV 高度同源的M 基因[29],可增加跨种感染风险、导致与哺乳动物致病力增强相关的A198T、Q234L(HA),L89V、G309D、T339K(PB2),D622G(PB1),N30D、T215A(M1),P42S(NS1)突变[19-21]。虽然野禽源H9N2 AIV 与家禽分离株有着几近相同的氨基酸变异位点,前者缺失PA 蛋白N383D 突变位点,如前所述病毒在天然宿主体内温和复制,不表现临床症状,因此野禽源AIV 无该突变,可能是病毒对天然宿主的适应性改变。这些研究结果提示,持续监测野禽携带AIV 的变异情况,并分析其与家禽源AIV 的相似性,探究分离株的跨种属传播能力,可以为禽流感综合防控供基础数据。
